
Jeff Jones, Program Manager at Sewtec Automation, provides an insight into how these changes are being introduced and what they mean for the production and packaging of the industry’s products.
The concept of Unique Device Identifiers (UDIs) has long gone hand-in-hand with the production and distribution of MedTech products in Europe and the US.
The US Food and Drug Administration (FDA) has mandated the inclusion of UDIs on products marketed in the US since 2013; and, with many companies supplying products into North America doing the same in Europe, coded identifiers are also a common sight on this side of the Atlantic.
Historic use of UDIs
In short, UDI regulations require the primary packaging of every medical device to carry a code displaying the product’s serial number and information relating to the manufacture of the device. As a result, the end user — whether a pharmacist or a consumer — can access key information about a specific product should they need to. It also provides authentication that a product is legitimate, rather than counterfeit.
The main drawback of the FDA’s UDI regulation is that the system does not enable a product to be tracked through the whole supply chain. However, thanks to a new set of proposals put forward by the European Union (EU), that is about to change, bringing with it a much more comprehensive set of track and trace requirements for manufacturers.
Introducing the new European Medical Device Regulations 2017/745
First proposed in 2017, the new EU Medical Device Regulations (MDR) 2017/745 proposes a new type of serialisation comprising a UDI for every product pack, which is then aggregated at every stage of the supply chain.
Under these new requirements, all UDIs will be stored on a centralised European database, which is set to be created and controlled by the European Database on Medical Devices (EUDAMED).
The key difference here is the aggregation. For the first time, it is made possible for a product to be tracked at every step of its journey from manufacture to consumer transaction. For example, it may be that a primary pack is placed within a shelf display carton before being packed in a case and then onto a pallet.
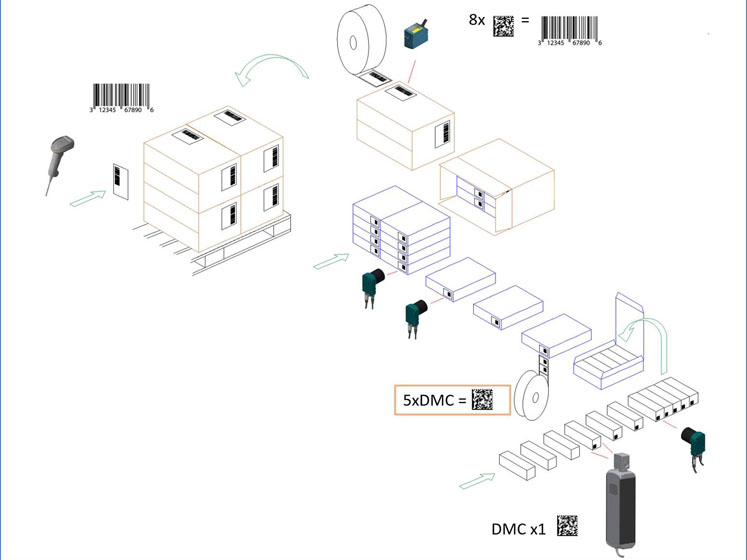
Under previous regulations, it would have been impossible to check how far along the supply chain the product was unless you had the identification of the pallet or case. With the new proposals, however, it’s possible to track any product to its most recent process.
The proposals may initially appear to add complexity to the supply chain and demand greater sophistication from packaging lines, However, these complexities also bring with them a number of benefits.
First, the drive to suppress counterfeit products remains high on the industry’s agenda and this will be bolstered by greater traceability. Secondly — and arguably of greater direct benefit to a manufacturer’s bottom line — the new regulations aim to simplify the product recall process.
Should there ever be an issue with a specific batch of medical devices, for example, the new system will make it easier to pinpoint the specific batch affected rather than requiring the manufacturer to recall all stock.
MDR 2017/745 was originally set to come into force on 26 May 2020 but is currently delayed in light of the coronavirus pandemic. Although we may not know a specific date for the implementation of this new regulation, it is certain that manufacturers need to prepare to offer a greater level of serialisation and aggregation than they may have done previously.
Reviewing any existing packaging and labelling lines to establish their level of compliance with the new requirements is certainly an advisable first step for those yet to do so.


