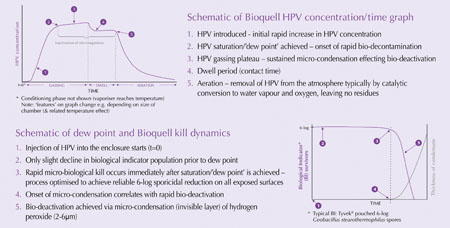
As companies look to develop large molecule products using targeted delivery methods, different quality control measures are needed. Tim Flanagan, Bioquell, looks at how bio-decontamination and monitoring processes are being adapted to meet this change.
It is imperative that medicinal products are manufactured within a clean and sterile environment. The risk of biological contamination during the manufacturing process needs to be reduced to a minimum. This ensures patient safety, and maintains production efficiency by eliminating the time-consuming and costly process of investigating the causes of contamination and the controlled destruction of contaminated batches.
There are clear and complementary regulatory requirements and references to ISO 14644 standards for classification and monitoring of controlled environments in the manufacture of sterile medicinal and therapeutic products. Regulatory requirements in the US are detailed in the FDA Guidance for Industry in Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice, United States Pharmacopeia (USP) including Microbiological Evaluation of Clean Rooms and Other Controlled Environments USP <1116>. In Europe the EU Good Manufacturing Practice – EU GMP annex 1 applies.
Sterility tests are required during the production validation process, as well as for routine quality control and detecting major contamination in a batch. Sterility testing can be an extremely difficult process and open to errors. Protocols must be designed and executed so as to eliminate false results. With the increasing number of medicinal and therapeutic products that have a biological profile and therefore cannot be terminally sterilised, there is also a greater requirement for aseptic processing where both biological contamination control and sterility testing play a part in batch release.





