Accurate scale-up using a well thought out strategy is a significant aspect of developing a robust manufacturing process for a topical drug formulation. Development programmes often benefit from even modest financial and time investments in structured process development.
The value of these contributions is clearly demonstrated when the risks of costly batch failures and project delays are mitigated. Although this strategy is applicable to both products based on new chemical entities (NCEs) and generic drug products, it is unfortunately often overlooked.
As the volume of generic topical products increases, it’s important for manufacturers to understand any gaps in the process development chain of the originator product. Developers need to be aware of the possibility that critical process parameters may not have been well defined, resulting in suboptimal products.
A Quality by Design (QbD) approach enables the development of a robust control strategy, the outcomes of which remain with a product throughout its entire lifecycle. This strategy derisks processes from initial scale-up (for clinical or toxicology batches) to commercial production and extends to plant-to-plant transfers further in the future.
A robust process control strategy can be derived by combining six-sigma tools with QbD. Integrating quality into the development process helps regulatory authorities to ensure that a consistent manufacturing process has been established.
This, alongside reinforcing a product’s commercial viability, are just two benefits of an integrated QbD approach to the product owner. In addition to using experimental design to challenge critical process parameters (CPP), there are a number of different six-sigma tools that add to the power of QbD. These include benchmarking, input-output diagrams, voice of the customer, evaluation criteria and failure mode effect analysis (FMEA).
Application of QbD to topical formulations
When developing semisolid products, defining a control strategy based on critical material attributes (CMAs) and CPPs is of particular importance. By carefully considering each aspect of the target product profile and any technical and scientific constraints inflicted by the active pharmaceutical ingredient (API), excipients and their combination, the composition of semisolid products is derived.
It is understood that each of these attributes can have a profound effect on the resultant safety and efficacy of the developing formulation.
However, it is also important to recognise that they can potentially dictate the manufacturing process, which is especially applicable in areas such as dissolution, mixing parameters and heating or cooling processes. There is a clear relationship between the safety, efficacy and composition of topical pharmaceutical products such as creams, gels, foams and ointments.
Nevertheless, the effect of the association between clinical performance and a product’s microstructure is often undervalued — a relationship that is highly dependent on manufacturing process parameters.
It is imperative that any development programme focused on product optimisation considers a broad range of processing parameters to guarantee the consistent manufacture of the pharmaceutical products to the required specification.
As the complexity of the topical formulation increases, so too do the processing parameters. And, as the complexity of these various processing requirements increases, so does the influence of the CPP on control strategy and the complexity of a scale-up programme.
An example of these more complicated topical pharmaceutical products is creams, owing to the need for defined processes and the order of addition, speed of mixing and stirrer timings as a result of combining two immiscible phases.2 These defined processes often extend to creating predetermined controlled heating and cooling rates.3
Generation of CQAs
The quality target product profile (QTPP) is a key tool; used from the start of any pharmaceutical development, it ensures that the broad objectives of a project are captured and clearly understood. The QTPP covers multiple aspects, from patient and prescriber requirements to the attributes needed to ensure a commercially viable, safe and effective treatment.
The QTPP should be referenced when determining the critical quality attributes (CQAs) — in combination with the likely impact of material attributes and process parameters on product quality.4 In new product development, using competitor products as a benchmark provides a useful guide to the QTPP as it can highlight any commercially relevant differentiators. This is particularly useful in the generic drug market.
Voice of the customer (VOC) is a six-sigma tool that allows both the end user and the company sponsoring the development to clarify their product requirements. VOC is a key element in the compilation of the QTPP. In regard to topical product development, this is paramount; these end user voices are often captured through online patient groups.
Other key opinion leaders (KOLs) such as prescribers and investors are also critical voices in the development of topical products; here, relevant information can be captured efficiently using surveys. VOC and the QTPP play a crucial role in the determination of CQAs.
Incorporating risk management
Risk identification and mitigation are important elements of any efficient process development programme. Whether creating a control strategy for NCEs or generic drugs, a well-recognised route to producing products of appropriate quality is through a combination of the successful deployment of FMEA as a risk management tool and an appropriate experimental design.
FMEA is a step-by-step process used to estimate any potential risks that could arise from all possible failures in the design or processing of a topical product. The FMEA defines where efforts should be focused and the most suitable process controls in the case of any of these potential risks occurring.
Many liquid and semisolid topical pharmaceutical products are highly complex systems, often involving multiple phases (such as water and oil emulsions) with a defined range of droplet sizes. As a consequence, failures can occur in a number of processing areas, such as drug content heterogeneity, consistency or physical, chemical and microbial instability.
For topical pharmaceutical products, core elements such as efficacy, uniformity of dose and safety are heavily reliant on the stability, homogeneity and ease of use.
Focusing on CPPs
To create the CPPs, the relationship between the processing parameters, the CMAs of both the API and the excipients, and the CQAs of the product must first be established. The six-sigma input–process–output (IPO) diagram (Table I) provides a succinct way of expressing the process parameters. The IPO diagram highlights the unit operations and which parameters should be investigated during the manufacturing process.
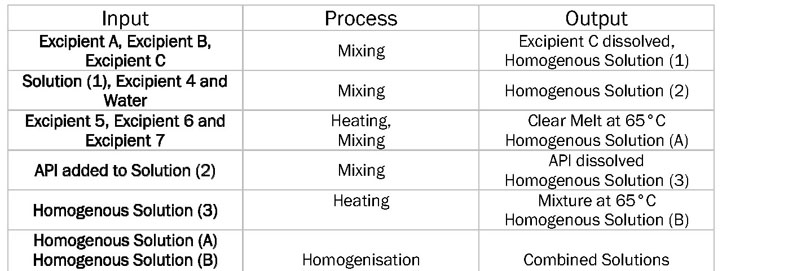
Table I: Input–process–output (IPO) diagram showing the unit operations of a sample manufacturing process
Derived from both the outputs of the FMEA (Table II) and the IPO diagram is the first design of experiment (DoE) in the form of a prescreening. There are multiple objectives for this stage, including
- confirming the output from the FMEA
- uncovering any interactions between key parameters
- distinguishing any CPPs.
FMEA allows the operator to effectively elucidate the most critical areas of the process, which might require further experimental investigation, whereas the DoE works to statistically and quantitatively validate this. The amalgamation of the experience and expertise of the technical project team and efficient experimental design software are essential for this approach.4
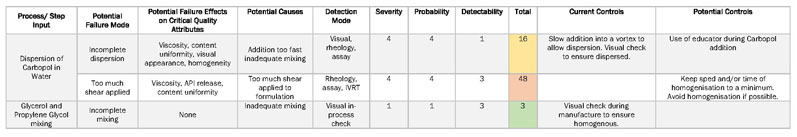
Table II: Failure mode effect analysis (FMEA) is used to highlight unit process operations that pose the most risk to product quality (IVRT is in vitro release testing)
Key factors in the design of experiments
During the prescreening and full DoE in topical formulation development, it is critical that experiments are conducted using equipment that is representative of that which will be used at a larger scale. This approach works to safeguard the derivation of meaningful qualitative CPP data and therefore the robustness of the resultant control strategy.
To allow for the control of all typical processing parameters, MedPharm uses IKA LR 1000 lab reactors. The basis of any future scale-up work and technical transfer activities are formed by understanding the influence of scale on the CPP from high-quality experimental work conducted on a small scale.
As previously mentioned, creams are highly complex topical drug products that typically require twelve experiments covering two to four CPPs to be done in a prescreening study in preparation for the manufacture of toxicity or clinical batches. The final number of experiments may need to be expanded and this is dependent on the outputs and associated risks identified in the prescreening study.
To gain a clearer understanding of design and control space boundaries, DoE — in both prescreening and the full studies — should attempt to push the topical product to failure. This tactic helps to identify and avoid any problems that could arise as a result of conservative experimentation and misinterpretation of the boundaries between success and failure, limiting the design space to the knowledge space (Figure 1).
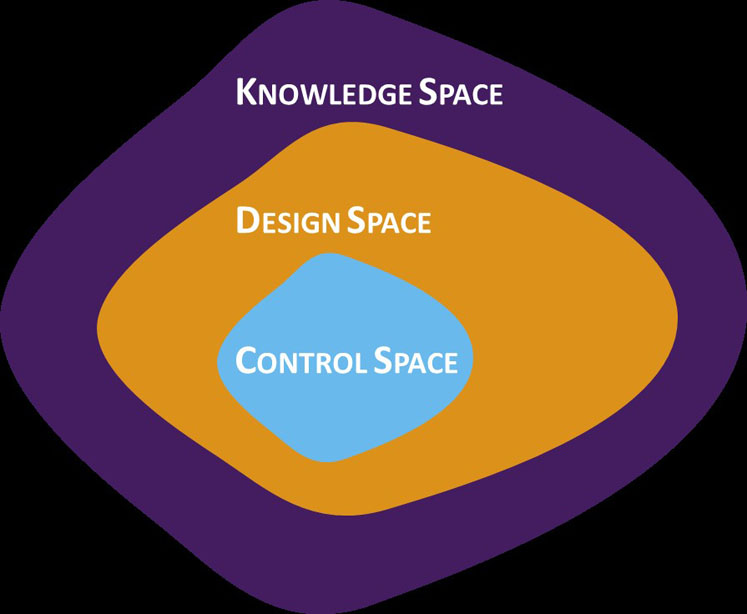
Figure 1: A graphical representation of the relationship between knowledge space, design space and control space
Accompanying these strategies is a third crucial factor: the key process output variable(s) (KPOV). As defined by six-sigma, KPOVs determine success and it’s critical to establish whether the method employed to measure the KPOV will detect critical failure. If this is not the case, a mitigation plan is needed as any experimental work will only produce outputs as accurate as the analytical method allows.
The evaluation of the quality of topical pharmaceutical products employs the use of an array of analytical techniques, which range from well-established methods such as high-performance liquid chromatography (HPLC) and viscosity testing to more sophisticated methodologies, such as rheological evaluation and in vitro release testing to ensure that there is no change in the release of the drug from the formulation.
The design space is defined as the multidimensional combination and interaction of input variables and process parameters that have been demonstrated to provide assurance of quality.5 It satisfies the QTPP and CQAs for the product and provides the boundary of any process parameters to which a product can be made.
It is important to stress that the knowledge space is not a hard boundary and that defined process inputs are just as important as the measured output.6
Interpretation of the DoE using statistical techniques such as analysis of variants (ANOVA) plots, which show the mean and distribution around the mean, should clearly show the significance of any parameter and any key interactions between parameters and any associated variability.
With an increase in the sophistication of many experimental design software packages (such as SPSS, Minitab, JMP and Design Expert), it’s easier than ever to highlight statistically significant effects across a range of parameters and present them in a concise graphic that decision makers find easy to understand.
Regulatory bodies have allowed for flexibility in any future manufacturing processes by concluding that changes made within the design space are not considered to be regulatory ones.7 Therefore, understanding more from the outset about the boundaries of success and failure is a critical element in creating the foundations for a good control strategy in the eyes of the developer.
The result is a mitigation of many negative impacts during a product’s lifecycle as a result of excipient supplier differences or changes in specifications or site of production.

Ensuring a robust process
The control space signifies the range of CPPs within which the output can be ensured to meet the CQAs and target specification at all times. When crossing from the design to the control space, the use of DoE is once again essential. This step is typically informed by the data from previous DoE work and involves two to five full or fractional factorials or another surface response design. The engineering statistics handbook provides a useful online guide to DoE.7
Ensuring robustness of the formulation is undoubtedly a result of the control space for the CPP sitting well within the boundaries of the design space and sufficiently away from the edge of failure. An optimisation design provides interpretations and conclusions to show the settings that would be the most desirable to ensure that the QTPP and CQAs are met for a topical product.
The production of a confirmation batch allows the developer to demonstrate that any predicted values are indicative of response values from the DoE.
Conclusion
The use of a methodical QbD approach throughout the formulation development of topical products provides a robust platform to establish the correct design space for process development. The integration of this approach with QbD provides a reliable control strategy for manufacturing. As previous development programmes can evidence, missing this critical step in the production of complex liquid and semisolid topical products cannot be underestimated.
The incorrect implementation, or lack thereof, of QbD in topical formulation development can lead to poor processing and physical instability, which consequently has direct impacts on development timelines, product performance and, ultimately, patients.
With regard to complex topical products, combining QbD with industry experience, six-sigma tools and DoE ensures that scale-up can be conducted to a high-quality standard with minimum risk.
Above all, industry leaders who are familiar with using a well thought out QbD-based process development strategy can combine this with in-depth knowledge of their products and processes to derisk topical formulation development, fundamentally saving both time and money.
References
- www.marketsandmarkets.com/Market-Reports/topical-drug-delivery-market-124871717.html.
- M.E. Aulton and K.M. Taylor, Eds., Aulton’s Pharmaceutics, Fourth Edition: The Design and Manufacture of Medicines (Elsevier, Amsterdam, the Netherlands, 2013) p 449.
- M. Lowenborg, “Challenges in Topical Drug Manufacturing,” Pharm. Tech. 36(11): www.pharmtech.com/challenges-topical-drug-manufacturing-0 (2012).
- R.K. Chang, et al., “Generic Development of Topical Dermatologic Products, Part II: Quality by Design for Topical Semisolid Products,” AAPS J. 15(3), 674–683 (2013).
- ICH, Q8 (R2) Pharmaceutical Development: www.ema.europa.eu/en/ich-q8-r2-pharmaceutical-development (August 2009).
- M. Adams, “Quality by Design and Design for Six Sigma,” presentation at the ISPE Breakfast Seminar (Toronto, Canada, May 2009).
- www.itl.nist.gov/div898/handbook.






