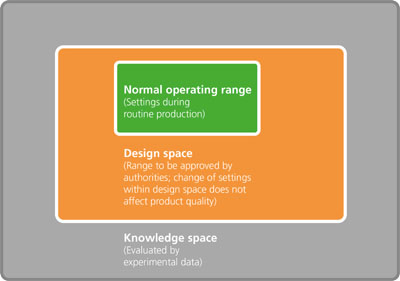
Now, however, we are seeing an increased emphasis on tackling quality upfront in the pharmaceutical manufacturing process. This means introducing quality as a consideration during the R&D phase rather than waiting until the product is designed and then thinking about how quality will be maintained. After all, quality cannot be tested into a product, it should be there by design.
An approach widely adopted in other industries that is gaining traction within the pharmaceutical industry is Quality by Design (QbD). Combined with process analytical technology (PAT), QbD is driving the move away from empirical methods towards a systematic, data-driven approach.
Not only are these twin approaches highly recommended by regulatory authorities, they also result in the development and manufacture of better products with improved efficiency and a reduction in late-stage quality issues. It’s becoming increasingly clear to the pharmaceutical industry that the advantages significantly outweigh the investment.




