Reasons for this include the challenges involved in penetrating and surviving in the notoriously immunosuppressive environment of a solid tumour.
One approach to solving this problem is to use genetic engineering to increase the persistence and proliferation of a therapeutic cell in challenging microenvironments. Although such alterations can be introduced into immune cells, such as T cells, when extracted from a patient, this autologous process is costly and time consuming.
Thus, focus has switched to the development of allogeneic universal cells: off-the-shelf cell therapies that have different capabilities introduced through gene editing. The drawback is that these cells will also need to adopt a “stealth mode,” allowing them to evade the body’s immune response to a perceived foreign invader.
Achieving an allogeneic universal cell with these properties requires multiple gene engineering events; and, although there are several gene editing techniques that could achieve this, the newcomer on the scene, base editing, is well suited to drive the next evolution of cell therapy for solid tumours.
Immunotherapy as a way to treat cancer is not a new concept: antibodies have met with success and revolutionised survival rates for some cancer patients for more than 20 years. The initial idea that an immune response can aid or even facilitate remission dates from 1893 and the work of William Coley.1
More recent data from mouse models indicate that tumour cells do initially provoke an immune response, possibly owing to mutated proteins present in tumour cells providing new “antigens,” which are identified as non-self by the immune system.

Emily Anderson
However, as the tumour develops, this immune response becomes less robust and is eventually ineffectual at stemming the proliferation of the tumour cells. Recent efforts to try and provoke a patient’s immune response into “rerecognising” tumour antigens using immune checkpoint inhibitory antibodies have also achieved clinical success.
These antibodies work in part by preventing proteins on the surface of the tumour cells interacting with proteins on the surface of immune cells, such as T cells.
These protein–protein interactions naturally limit the extent and duration of an immune response, but the expression of these inhibitory proteins on the surface of tumour cells is just one of the immune suppressive mechanisms that a patient’s immune cells encounter in an established tumour.
Cancer treatment using CAR-T cells
Hard on the heels of checkpoint antibodies has been the successful development and use of chimeric antigen receptor (CAR)-T cells (Figure 1). T cells from a patient are harvested and genetically modified to express a CAR known to target a protein on the surface of the tumour cells.
These cells are then reinfused into the patient. As these engineered CAR-T cells are derived from the patient, they are autologous and are not seen as “foreign” by the patient’s immune system. Although successful, the autologous approach is laborious and difficult to scale, requires patient-derived T cells that can survive the engineering process … and is expensive.
As CAR-T cells and other immune cell-based CARs appear to have broader efficacy for the treatment of cancer, there is a need to develop a more robust and scalable approach to generate CAR-immune cells.
One option is the use of non-self or allogeneic T cells, which could be engineered with different CARs and stored in bulk ready for distribution to hospitals to treat patients, similar to any other approved off-the-shelf therapeutic.
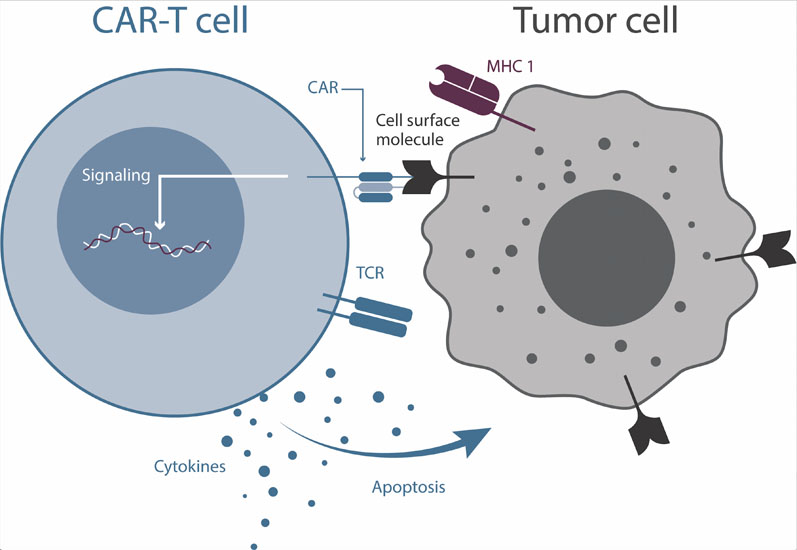
Figure 1: Cartoon depiction of a CAR-T cell interacting with a cancer cell
In addition, it is feasible that such allogeneic CAR-immune cells could be derived from “universal donor” cells that are genetically modified to evade detection (stealth cells) by a patient’s own immune system.
Allogeneic bone marrow transplants have been used for many years; however, the allogeneic approach faces significant challenges, such as the risk of graft-versus-host disease and a lack of durable allogeneic cell survival.
Despite these challenges, in April 2019 the US Food and Drug Administration (FDA) approved the first allogeneic CAR-T for investigational use in patients with multiple myeloma, and more are expected to follow. One additional advantage of allogeneic therapeutic cells is that their manufacture could be less expensive than the current autologous alternatives.
To give some perspective on the cost difference, the current pricing of the autologous KYMRIAH and YESCARTA treatments are $373,000 and $475,000 per patient, respectively. In comparison, a study in 2018 predicted that an allogeneic approach could reduce this by more than 50-fold to around $7500 per patient.2
So far, regulatory approval of CAR-T therapies has been limited to liquid or blood-based cancers.
Although currently in clinical trials, CAR-T cells targeting the more common solid tumours face additional hurdles that are less prevalent in liquid tumours.
Some of these hurdles, such as increasing survival and reducing the susceptibility of CAR-T cells to immunosuppressive signals, could be lowered or removed using gene editing to further improve the CAR. CRISPR-Cas9 is an accessible approach to gene editing that surpasses the throughput of previous technologies, as the Cas9 nuclease can be programmed with a synthetic RNA sequence to target almost any genomic location of interest.
However, the application of first-generation CRISPR-Cas9 techniques has only allowed for the programmed formation of DNA double-stranded breaks (DSBs). After this event, the cell is left to its own devices to repair the break, most often via error-prone non-homologous end joining, creating a spectrum of small insertions and deletions that can result in gene disruption and effective knockout.
Notably, in the case of engineered allogeneic T cells, edits in multiple genes will be required to create the universal, supercharged stealth cell. Creating simultaneous DSBs in multiple genes of one cell could result in unwanted translocations in which two DSBs on different chromosomes fuse.
Such translocations are likely to be deleterious for the cell or, more rarely, could form fusion genes that are oncogenic drivers. This likelihood is exacerbated with multiple edits as there are, potentially, exponentially more free DNA ends available for erroneous repair with each additional gene cut.3
Base editing to the rescue
A promising approach to enabling multiple gene edits would be one that does not create DSBs. Enter the second generation of CRISPR-Cas technology: base editing (Figure 2). This approach to gene editing involves a partially deactivated Cas9, which nicks one strand of the DNA, but does not cut both strands, thus avoiding potential translocation events.
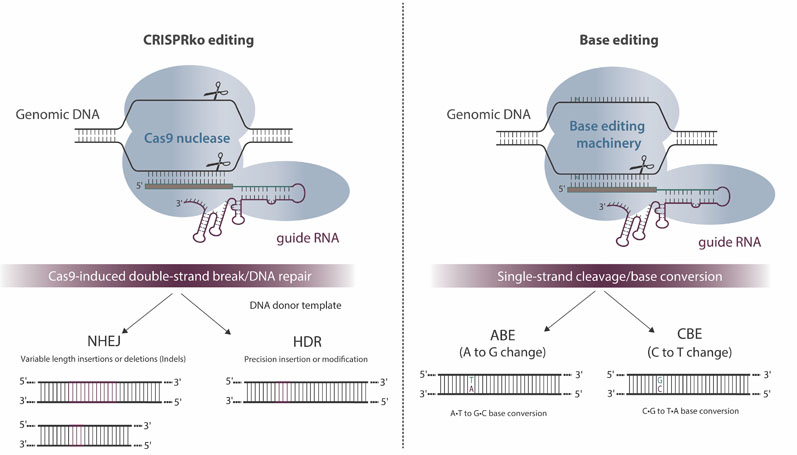
Figure 2: Overview and potential outcomes of CRISPR-Cas9-mediated genome editing and base editing
Combine this “nickase” Cas protein with a cytidine or adenosine deaminase and the cell can be coaxed to repair deaminated bases through the mismatch repair pathway (thereby converting C bases to Ts or A bases to Gs) with no DSBs required. This technology was first reported in peer-reviewed papers published by researchers at Harvard University.4
Others, such as Rutgers University, have also developed base editing platforms.5 Base editing can be used to specifically knock multiple genes out through the introduction of stop codons or splice site disruptions, and should substantially reduce the risk of genomic alterations (such as translocations).
Engineering stealth cell therapies will most likely require multiplex editing using a safe technology that limits the potential to cause damage elsewhere in the genome. To reach this goal, many challenges must be overcome, including
- the selection of gene targets in the right combination to improve the fitness of CAR-immune cells
- the evolution of ever-better base editing modules and configurations of these molecules
- delivery and efficacy in T cells (and other immune cells that may be harnessed in the future for cell-based therapy)
- the daunting challenge of production of GMP-grade proteins and RNA components.
These challenges are not insurmountable, however, and base editing provides a compelling route forward.
References
- W.B. Coley, “The Treatment of Malignant Tumors by Repeated Inoculations of Erysipelas: With a Report of Ten Original Cases,” American Journal of Medical Sciences 10, 487–511 (1893).
- M. Jenkins and S. Farid, “Cost-effective Bioprocess Design for the Manufacture of Allogeneic CAR-T Cell Therapies Using a Decisional Tool with Multi-Attribute Decision-Making Analysis,” Biochem. Eng. Journal 137, 192–204 (2018).
- G. Giannoukos, et al., “UDiTaS, A Genome Editing Detection Method for Indels and Genome Rearrangements,” BMC Genomics 19, 212. doi: 10.1186/s12864-018-4561-9 (2018).
- A.C. Komor, et al., “Programmable Editing of a Target Base in Genomic DNA Without Double-Stranded DNA Cleavage,” Nature 533(7603), 420–424 (2016).
- https://horizondiscovery.com/en/news/2020/Horizon-Discovery-to-provide-access-to-novel-base-editing-technology.