The new chapter USP <665> addresses the issue of production equipment and its potential influences on patient safety. What does this mean for resins made of polymeric-based bead materials? And how can manufacturers assess the risk profiles of commonly used resins in drug manufacturing? A detailed study from Merck provides the answers.
In its GMP General Guidelines, the US FDA states that “equipment shall be constructed so that surfaces that contact components, in-process materials or drug products shall not be reactive, additive or absorptive so as to alter the safety, identity, strength, quality or purity of the drug product beyond the official or other established requirements.”1
The US Pharmacopeia (USP) addresses the issue of production equipment and its potential influences on patient safety in the upcoming chapter USP <665>, titled “Plastic Components and Systems used to manufacture Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Products.”2

Maren Apostel
The current draft covers requirements for a broad range of polymeric components and systems in drug manufacturing. As it could add impurities to the final drug product, any component used during the later stages of the manufacturing process is considered to be a risk. As chromatography is an indispensable step in many drug production processes, it makes sense to apply this approach to resins made of polymeric-base bead materials.
Chromatography resins in the manufacturing process
Separation and purification of the molecule from a complex feed stream is among the most important steps in biopharmaceutical manufacturing. As it delivers the necessary purity and yield of the molecule from upstream processes, chromatography is essential.
Moreover, chromatography enables robust and cost-efficient manufacturing — a vital aspect given the highly regulated environment in which accelerated timelines and efficiency are crucial.
The chromatography steps used in biopharmaceutical production vary considerably, depending on the respective molecule type, chromatography technique and, most importantly, the process stage. Most purification systems include multiple chromatography steps (Figure 1) to achieve the level of purity needed for downstream applications.
Selecting the most appropriate chromatographic methods in the right sequence is essential to optimise the purification process. Moreover, finding the ideal procedure to individually pack the chromatography columns and evaluate the risk of extractables in connection with the polymeric chromatography resin is essential.
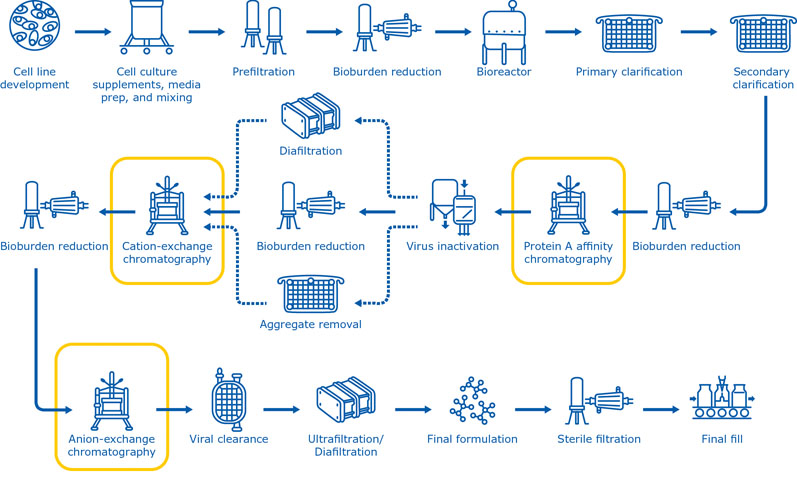
Figure 1: The manufacturing process for monoclonal antibodies typically includes multiple chromatography steps
New requirements for plastic components and systems
Polymeric components come into contact with the drug substance at various points in the manufacturing process. Such interactions can introduce compounds called process equipment-related leachables (PERLs), which are organic or inorganic compounds that passively migrate into drug substances or products and have the potential to alter their key quality attributes.
Future USP <665> guidance defines new requirements to make sure that PERLs do not compromise patient safety.
The chapter includes specifications regarding how to assess polymeric components for extractables, the chemical entities that can be released by a process system under certain conditions — such as high heat or pressure — as well as powerful acids, organic solvents and alkaline solutions. These conditions represent a worst-case scenario and do not occur in a standard manufacturing process.
Chapter USP <1665> provides additional information about the characterisation of polymeric components used in the manufacturing process.3
The purpose of these requirements is to differentiate between manufacturing components or systems that should be fully evaluated for organic extractables and those that require only minimal testing. Although the specifications of USP <1665> offer general guidance for the testing requirements, process owners are ultimately responsible for performing and verifying their risk evaluation.
From initial to risk assessment
As defined in the upcoming USP <665>, the initial assessment determines whether a plastic component or system is suitable for its intended use without further characterisation.
The first and second steps of the initial assessment evaluate whether there is meaningful contact between a component and a process stream, as well as the physical state of the process stream.
If the assessed component is isolated from the process stream, does not come into contact with a liquid or if a comparator system can be established, then no further characterisation is required (Figure 2).
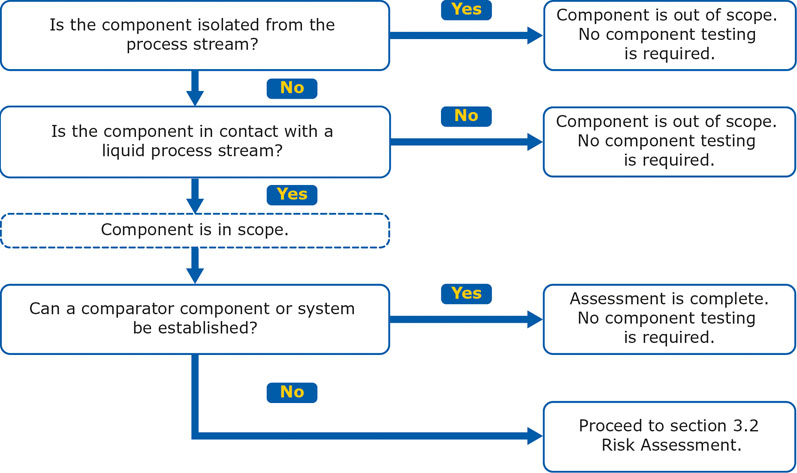
Figure 2: Initial assessment for a plastic component or system according to draft USP <665>
According to USP <1665>, the risk assessment for in-scope components employs a risk evaluation matrix that assigns three risk levels and considers four aspects: duration of contact; temperature of contact; chemical nature of the process stream; and nature of the component material.
The four aspects for the different dimensions are summarised in a final risk level (low, moderate or high). Based on mitigating factors, the characterisation level can be adjusted by implementing additional manufacturing steps or adapting clinical use.
A high risk level can be reduced, for example, by considering the removal of PERLs via one or more manufacturing steps and modifying the clinical use of the manufactured drug product.
Adapting upcoming USP <665> approach to chromatography resins
Components used late in the process generally pose a higher risk. At this stage, few opportunities remain to remove PERLs or other impurities. Chromatography steps are tailored to specific molecules and processes.
In addition, the exact downstream location of these steps can vary considerably. Therefore, risk profiles of individual chromatography resins must be evaluated in connection with specific process steps and extractable profiles can be used to support a comprehensive risk assessment process.
Merck approached this task by systematically assessing its portfolio and determining extractables profiles for bulk chromatography resins.
Chromatography resins are not used in a dry state; they are delivered in a storage solution (20% ethanol/150 mM/L NaCl, for example) that needs to be removed prior to use in the downstream process.
The resins must be washed in a pretreatment step, replacing the storage solvent with water or buffer solutions, followed by a packing procedure. Therefore, slight adjustments in sample treatment and analytical procedure were necessary.
Worst-case scenario as basis
By applying the USP <1665> risk assessment matrix for affinity and ion exchange steps in a typical monoclonal antibody (mAb) process, the chromatography steps would be considered as low to moderate risk components.
However, Merck decided to apply the full testing standards to support a worst-case approach. Manufacturing processes are highly specific and chromatography steps can be located close to the final drug.
Accordingly, data was generated for three extraction solvents — pH 3, pH 10 and 50% ethanol — as is required for a high-risk level component according to the USP <665> draft.
The pretreatment procedure included washing with 10 bed volumes of water. Extraction buffers and solvents were used in a higher concentration to allow for the dilution water in the resin.
The extraction was performed within 24 hours and at 40 °C. Most detected components could be traced back to the base matrix or to additives deriving from the packaging material.
Overall, the data showed that the probability of chromatography resins adding significant risk to the safety of a drug product can be considered to be low. A separate worst-case approach in line with ICH Q3D was used to determine elemental impurities.
Supporting customer risk assessments
What is true for chromatography resins cannot simply be assumed to apply to the entire manufacturing process. Given the high complexity of the matter, open and structured communication between drug manufacturers and chromatography resins suppliers is essential and can help to ensure a successful and comprehensive risk assessment.
In addition, suppliers should actively contribute to the drug manufacturer’s risk assessment process by following established guidelines, fulfilling information requirements and providing documentation in a timely manner, ideally bundled in packages and transmitted electronically.
The Emprove Program was designed to simplify this process, help drug manufacturers to overcome these challenges and support them in supplying the required documentation.
The Chromatography Dossiers include general information and product-specific data and supporting process optimisation efforts, as well as extended and safety risk assessment, including extractable profiles according to the USP <665> requirements.
References
- US FDA, 21 Code of Federal Regulations, Part 211, “Current Good Manufacturing Practice for Finished Pharmaceuticals,” www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=211.
- USP <665> Plastic Components and Systems Used to manufacture Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Products (1 September 2020).
- USP <1665> Characterization of Plastic Components and Systems Used to Manufacture Pharmaceutical Drug Products and Biopharmaceutical Drug Substance as and Products (1 September 2020).
Emprove is a registered trademark of Merck KGaA, Darmstadt, Germany