During the early phases of oral dosage form drug development, it’s not uncommon to only have a limited supply of a given active pharmaceutical ingredient (API).
This is because its synthetic route is unlikely to have been optimised for large-scale production and the API is only being produced at laboratory scale.
This limited API supply makes it challenging for formulation scientists to gather sufficient meaningful data to select the most promising candidates to advance into in vivo studies with the goal of selecting a clinical candidate.
As a drug development programme evolves from molecule discovery to animal pharmacokinetics (PK), animal toxicology studies and, ultimately, first-in-human studies, it is important that the formulations used provide adequate and consistent exposure of the drug to the patient to collect high quality data.
A traditional preclinical development approach may only afford limited preformulation and in vitro results, so numerous prototypes may be needed in initial animal PK studies to determine the ideal formulation for the compound.
This is often an iterative process and requires multiple rounds of formulation adjustments and PK studies to reach a conclusion, which is time consuming and requires greater quantities of API.
To optimise API usage for in vivo studies, it is often beneficial to have a preliminary understanding of the metabolism profile and absorption potential of the compound.
In silico modelling that can predict absorption, distribution, metabolism and excretion (ADME) parameters using the compound’s molecular structure is a good starting point, and models can be enhanced and refined by the inclusion of data from in vitro studies.

Tests using liver microsomes and/or a hepatocyte cell line can offer insights into metabolic susceptibility by human liver CYP450 enzymes. Compound solubility in biorelevant media such as fasted state simulated intestinal fluid (FaSSIF), fed state simulated intestinal fluid (FeSSIF) or simulated gastric fluid (SGF), along with an estimation of the intestinal permeability from Caco-2 cells, provide data on oral absorption potential.
The biorelevant solubility measurements and predicted permeability can be fed into the Developability Classification System (DCS) to provide an estimate of oral formulation development risk and guide formulation development further.1
Of the three key factors that determine oral API bioavailability, solubility, metabolism and permeability, it is on a molecule’s solubility that formulators can have the most impact.
Compounds that are inherently resistant to hepatic metabolism and/or display high permeability have the greatest potential as oral candidates to advance in development.2,3
If solubility enhancement is the main challenge to be overcome for a given molecule, there are a number of in vitro techniques that can be used on limited quantities of API to ensure the correct formulation for further studies.
Common API sparing techniques used during solubility enhancement formulation activities include small volume dissolution, two-stage dissolution, liquid-liquid phase separation (LLPS), polymer screening studies, film casting, crystallising tendency and flux measurements.
Deciding on which in vitro sparing technique to incorporate into a development plan may depend on API availability, time and the enhancing technologies under evaluation.
Some methods, such as small volume dissolution, provide value for any formulation approach wherein kinetic or equilibrium solubility is an issue, as discriminating dissolution is a mainstay to predict in vivo performance.
Approaches that look at the drug’s propensity to crystallise, or the impact of polymers on the physical stability of amorphous forms, are usually reserved for amorphous dispersion studies of crystalline APIs.
Techniques that incorporate movement across membranes are useful when the formulation components may impact permeation, which is often, but not exclusively, the case with lipid-based systems.
Small volume dissolution
Small volume dissolution and solubility apparatus is commercially available and allows experiments to be done in small vessels — typically 10–20 mL — that are monitored using in situ fibre-optic probes.
The set-up has the advantage of limiting API consumption and affords better understanding of dissolution behaviour as API concentrations are measured every few seconds compared with minutes during typical full-scale dissolution experiments.

This type of apparatus is well suited for solubility studies when looking at kinetic solubility enhanced products, (for example, micronised materials) or amorphous solid dispersions (ASDs) of crystalline drugs.
Two-stage dissolution
Changes in the pH and the composition of the dissolution media can have a significant impact on API solubility. In the low pH of the stomach, weakly basic APIs typically exhibit a high solubility, but are at risk of precipitation when exposed to the higher pH environment of the small intestine.
Two-stage dissolution experiments investigating the behaviour of the API during this pH shift can be conducted using traditional USP dissolution apparatus or alternative instruments.4
During the pH shift, the API can remain in solution in a supersaturated state or precipitate either in the amorphous or crystalline form. In an ideal situation, the API would remain supersaturated in the small intestine for sufficient time to allow for absorption.
However, if the API precipitates at a high pH, then follow-up two-stage dissolution experiments are conducted to screen for polymers that can maintain the API supersaturation following the pH shift.
LLPS and polymer screening studies
LLPS is described by the formation of drug-rich colloidal species within a supersaturated aqueous solution of a poorly soluble API, and occurs when the concentration of the API exceeds its amorphous solubility.
The concentration at which LLPS occurs can be influenced by the media used (that is, FaSSIF, FeSSIF), along with the presence or absence of polymers within it.
However, this only provides an estimate of the amorphous solubility limit and additional experiments are required to identify which polymers can successfully maintain supersaturation of the API in the intestine, and will need to be incorporated into the amorphous drug dispersion.5
Again, small volume dissolution and solubility apparatus can be used to determine the ability of polymers to maintain API supersaturation in simulated intestinal fluid with only small quantities of API.
Additional experiments to evaluate the supersaturation behaviour at varying API-polymer ratios (for example, 25:75, 50:50 and/or 75:25), can then provide an initial estimate of the drug loading feasibility for an ASD formulation.
Figure 1 shows the theoretical dissolution behaviour of a poorly soluble compound as a crystalline drug, amorphous drug and an ASD. The crystalline drug displays low solubility, whereas the amorphous drug shows peak solubility but quickly drops.
The ASD achieves the spring effect seen with the amorphous drug and helps to maintain supersaturation in solution (parachute effect).
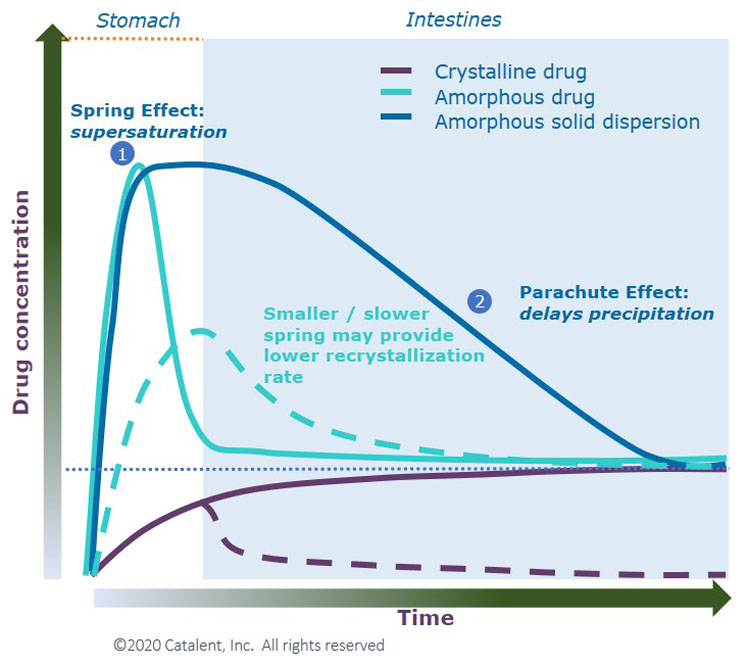
Figure 1: Spring and parachute concept to achieve high apparent solubility for insoluble drugs6
Film casting
Film casting is another technique used to screen for suitable polymers and estimate the drug loading capacity of an ASD formulation. Unlike LLPS techniques that focus on the maintenance of supersaturation in solution, film casting experiments provide an estimate of the physical stability of API-polymer mixtures.
In a typical experiment, the API and potential polymer are dissolved in a solvent and an aliquot of the solution is place on either a slide or into the wells of a 96-well plate where the solvent evaporates, leaving a film.
The film is then analysed by polarised light microscopy (PLM), differential scanning calorimetry and/or powder X-ray diffraction to look for signs of phase separation or API crystallisation.
The slides or plates are stored at 40 °C/75% relative humidity to study the effects of increased temperature and humidity on the film’s physical stability.
Crystallising tendency
PLM can also be used to study an API’s crystallising tendency, which again requires little API material.7 This technique is used to study an API’s inherent crystallisation properties and helps to identify molecules that exhibit a low crystallisation tendency, which might demonstrate their potential as good candidates for ASD formulation.
Small volume dissolution: flux measurements
The jejunum is the middle part of the small intestine, located between the duodenum and the ileum. Permeability across a jejunal-like membrane can be studied using instruments that measure the concentration of the API in solution across donor and acceptor compartments.
This can calculate the rate of flux across the membrane; and, whereas this does not specifically predict in vivo PK, these studies can rank drug candidate formulations in order with respect to their expected in vivo performance.8
These experiments are suitable for various formulations and can be most useful and insightful when investigating complex lipid formulations, whereby movement across membranes may be more directly impacted by the formulation.
Amid pressure to accelerate drug development and progress programmes into the clinic, creating drug formulations with the greatest chance of success from the earliest stages can reduce the risk of failure and time-consuming rework.
API supply is often limited in the early stages because of the scale of manufacture and also the cost of manufacturing, meaning formulation scientists need to capture as much data as possible with both careful planning and resource management.
References
- J. Butler and J. Dressman, “The Developability Classification System: Application of Biopharmaceutics Concepts to Formulation Development,” Journal of Pharmaceutical Sciences 99, 4940–4954 (2010).
- I. Gomez-Orellana, “Strategies to Improve Oral Drug Bioavailability,” Expert Opinion in Drug Delivery 2, 419–433 (2005).
- V. Thomas, et al., “The Road Map to Oral Bioavailability: An Industrial Perspective,” Expert Opinion in Drug Metabolism and Toxicology 2, 591–608 (2006).
- K. Suzuki, et al., “Relevance of Liquid-Liquid Phase Separation of Supersaturated Solution in Oral Absorption of Albendazole from Amorphous Solid Dispersions,” Pharmaceutics 13, 220 (2021).
- S. Hate, S. Reutzel-Edens and L. Taylor, “Insight into Amorphous Solid Dispersion Performance by Coupled Dissolution and Membrane Mass Transfer Measurements,” Molecular Pharmacology 16, 448–461 (2019).
- M. Karimi-Jafari, et al., “Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications,” Crystal Growth & Design 18, 6370–6387 (2018).
- A. Kapourani, et al., “Crystallization Tendency of APIs Possessing Different Thermal and Glass Related Properties in Amorphous Solid Dispersions,” International Journal of Pharmaceutics 579, 119149 (2020).
- K. Tsinman, et al., “Ranking Itraconazole Formulations Based on the Flux through Artificial Lipophilic Membrane,” Pharmaceutical Research 35(8), 161 (2018).