It’s therefore crucial for pharmaceutical companies and manufacturers to get up to speed with these latest requirements. In this article, Houri Simonian, PhD, Director of Analytical Solutions at SGS Life Sciences, will discuss in detail the requirements specified by global agencies for the testing and control of the formation of nitrosamines in the drug substance and product manufacturing process.
Background on nitrosamines
Nitrosamines are known environmental contaminants that are found in water and foods, including meats, dairy products and vegetables. But, after their recent detection in multiple drug products, they became a major concern for pharmaceutical companies, especially as nitrosamines are classified as probable human carcinogens.
ICH’s M7 mutagenic impurities guidelines classify this group of genotoxins as part of a group of high-potency mutagenic carcinogens referred to as the “cohort of concern.”
Their formation in pharmaceutical products was first highlighted in early 2018. Originally, high levels of N-nitrosodimethylamine (NDMA) were unexpectedly found to be above the acceptable limits in Valsartan, the angiotensin receptor blocker (ARB).
This led to further studies that highlighted their presence in multiple other drug products, including ranitidine, metformin and other ARBs. Now, manufacturers of both drug products and active pharmaceutical ingredients (APIs) must take measures to control the presence of nitrosamines by following the most recent regulatory requirements.
Regulations and testing
Since the nitrosamine impurities were found in various drug products throughout different markets, international regulatory authorities have partnered to share information and publish guidelines for market authorisation holders (MAHs). These recommendations include analytical methods to detect and identify nitrosamine impurities in drug products.
The guidelines also describe methods to identify the potential sources of nitrosamine contamination and formation, as well as approaches to control or eliminate these sources during the manufacturing process. Accordingly, a three-step risk assessment has been proposed.
- Step one: assess the risk of nitrosamine formation during drug substance (API) and drug product manufacturing (including products in development and approved or marketed products), taking into consideration potential root causes and sources of impurity formation
- Step two: perform confirmatory testing (in case a risk of nitrosamine formation is confirmed)
- Step three: implement and report changes made to prevent or reduce nitrosamine impurities.
Risk assessment (step one)
Nitrosamines can be formed during all three stages of the manufacturing process (Figure 1). For that reason, the risk of impurity formation should be evaluated from API synthesis to final product packaging. Common sources of nitrosamine formation are the solvents, reagents and catalysts used (especially amines) during API and drug manufacturing.

Figure 1: The three stages of drug production
Other sources include the presence of nitrites or nitrates in water and the contamination or cross-contamination of the equipment and starting materials used.
The FDA instructed manufacturers to complete risk evaluations and report the outcome by 31 March 2021. If an active substance was found to be at risk, MAHs should submit the step one response and proceed with step two confirmatory testing of the drug or product. If no risks are identified, a risk evaluation of the finished product should be conducted and the outcome submitted once a final conclusion is reached.
Confirmatory testing (step two)
If a risk of nitrosamine impurities is identified during the assessment step, then MAHs must conduct confirmatory testing on the products identified to be at risk and report any confirmed presence of nitrosamines as soon as possible. Acceptable limits to consider, based on a product’s maximum daily dose, have been proposed (Table I).
Although there is no requirement to use the published testing and analytical methodologies previously discussed, any method used should be quantitative, adequately sensitive, validated and conducted at a GMP-compliant facility. The LOQs and LODs mentioned by the regulatory agencies should be met to ensure that the method used is equivalent to published methods.
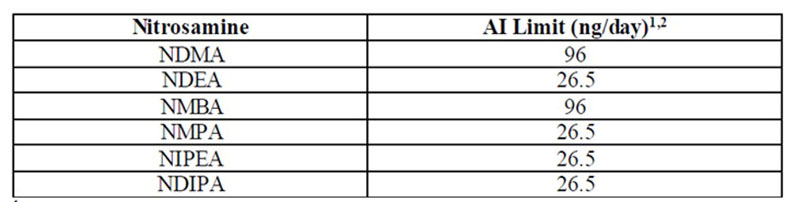
Table I: Acceptable intake limits
If a limit-based test is used, it must be accompanied by appropriate scientific justification in the risk assessment document, with evidence that there is no increase in the concentration of nitrosamine impurities with time. It’s recommended to use the drug product for the appropriate market for method validation and the choice of product strength should be described if the presence of multiple drug strengths is a possibility.
In addition, when developing analytical methods, there are multiple factors to keep in mind. Testers should consider any interferences caused by the presence of trace amounts of nitrosamines in testing materials such as water, plastic or rubber products. In situ formation of nitrosamines is also possible and should be accounted for, such as in the case of ranitidine in high-temperature conditions.
The EMA provides a table to analyse the results and identify the root causes of impurities.4 If nitrosamines are detected during testing, the root cause should be identified and stated in the report before moving to step three. If no impurities are detected, a report should be filed with (or be available to) the appropriate regulatory authority.
Implementing and reporting changes (step three)
If the confirmatory tests in step two validate the presence of nitrosamine impurities, MAHs should implement changes to prevent or reduce them in APIs and drug products. If one or more of the detected nitrosamine impurities are below the interim acceptable limit, then steps should be taken to determine their origin — as well as actions needed for respective batches.
Root causes should be determined and corrective and/or preventive actions should be implemented as well as a risk mitigation plan that ensures that levels will be consistently below interim acceptable limits at the end of the product’s shelf-life.
In addition to the guidance from regulatory authorities, compendial chapters have been proposed and/or submitted for publication.5,6 The compendial chapters are aligned with the guidelines in terms of the assessment and testing, as well as the recommended acceptable levels. Several methods using GC-MS, GC-MSMS or LC-MSMS have been published for the detection of specific nitrosamine impurities.
The general chapter also stipulates that alternative methods can be used to detect and quantitate nitrosamine impurities; however, these methods must be validated under actual use to meet respective performance characteristics criteria such as selectivity, sensitivity (LOQ) and recovery.
Nitrosamine control: a new reality
Under the new regulations, almost all drug products are mandated to undergo nitrosamine testing. Global regulatory agencies require all manufacturers and MAHs to perform and submit a proper risk evaluation before the specified deadlines. Even drug products that are planned for submission or have already been submitted require a risk assessment for the potential presence of nitrosamine impurities.
If the potential presence of nitrosamines has already been identified, the MAH is required to provide a risk assessment and a mitigation strategy, along with confirmatory testing plans or testing data. Only biological products that are free from any chemical excipients are exempt from nitrosamine testing for the time being.
The global approach of regulatory agencies to address nitrosamine formation shows the significance of this issue. The FDA, EMA and HC, along with agencies in many other parts of the world, are collaborating to share information in a concerted effort to align their requirements and actions globally.
References
- The AI limit is a daily exposure to a compound such as NDMA, NDEA, NMBA, NMPA, NIPEA or NDIPA that approximates a 1:100,000 cancer risk after 70 years of exposure. Appendix B (see reference 3) includes a description of the AI derivation for NDMA, which is an example of how the FDA applied ICH M7(R1) to set a limit.
- The conversion of an AI limit into parts per million (ppm) varies by product and is calculated based on a drug’s maximum daily dose (MDD) as reflected in the drug label (ppm = AI [ng]/MDD [mg]).
- www.fda.gov/regulatory-information/search-fda-guidance-documents/control-nitrosamine-impurities-human-drugs.
- www.ema.europa.eu/documents/template-form/step-2-nitrosamine-detected-above-acceptable-intake-new-nitrosamine-detected-response-template_en.xlsx.
- www.uspnf.com/notices/nitrosamine-impurities-gc-prospectus-20200424.
- www.edqm.eu/en/news/ph-eur-commission-adopts-new-general-chapter-analysis-n-nitrosamine-impurities.