Packaging rule changes in the new EU Clinical Trials Regulation (Regulation [EU] No. 536/2014) have caused concern among new drug sponsors by adding challenging operational hurdles to the new clinical approvals process. Joe Monteiro, Technical Account Manager at Essentra Packaging, talks to Dr Kevin Robinson to outline some of these changes, what they mean for those in the supply chain and what solutions are available.
KSR: So, what’s happening?
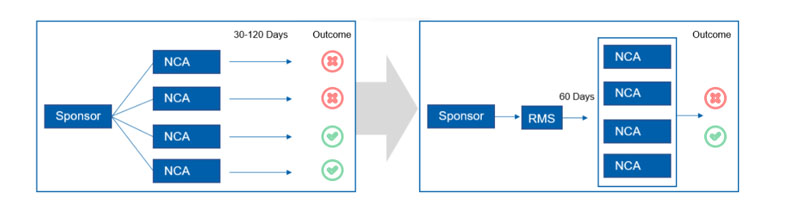
JM: On 31 January 2022, new regulations concerning clinicals trials will come into force throughout the European Union. The EU Clinical Trials Regulation exists to harmonise the approvals process across various member states whose own national regulations often didn’t align with each other, lengthening lead times on sometimes vital new medicines.
This legislation has repealed and replaced the Clinical Trials Directive that was brought into law in 2001 and provides an assessment and supervision process via a new Clinical Trials Information System (CTIS). The new process provides a “portal” that allows new medical products that require clinical approval to automatically be approved in all EU member states. The changes affect all new drug trials commencing after 31 January.
KSR: Isn’t this a good thing?
JM: The updates to the EU-wide regulations are extensive and we are optimistic that they will achieve the desired effect — except for a small clause concerning the packaging of trial medicines that has caused major concern from manufacturers and developers: Annex VI.
Approvals pipeline diagram
KSR: What is Annex VI?
JM: Although the new regulation’s measures are wide-ranging, the Annex VI clause specifically covers the critical information displayed on both primary and secondary packaging of medicines used in clinical trials, such as batch number and expiry date.
The main requirement that is causing concern is the requirement to label both the internal and external packaging. As an example, in a drug trial when the product is in a pill format, the necessary information must be affixed to the blister pack containing the pills themselves and the carton at the same time. Critically, if the information such as the expiry date changes, then the labelling on both the primary and secondary packaging needs to be updated before distribution to patients or trial participants.
KSR: Why is this a problem for drug sponsors?
JM: A regulation such as this works well for approved medicines that have progressed to the manufacturing stage. However, in the clinical trials sector, in which expiry dates, directions for use and reference numbers frequently change as a matter of course, this raises some significant operational challenges that will result in the unintended consequence of delays to clinical trials.
Drugs manufactured as part of clinical trials do not benefit from the same economies of scale as approved medicines and can be incredibly expensive on a per dose basis. It is this lack of scale that is the source of many of the challenges of meeting Annex VI requirements.
If indeed the labelled details do change, then a process of individually unpacking, relabelling and repacking needs to take place. Regardless of whether the drug developer has the necessary capacity to do this, time and cost is still added to the process.
KSR: Is labelling the only part of the process impacted?
JM: It’s not just the relabelling process that needs to be costed but the associated stages of production too, such as quality control, reworking of facilities, the possible need to find new suppliers and investment in new equipment. Plus, this doesn’t even consider the difficulties of trial drugs with high packaging demands.

To walk through a few examples, drugs with onerous cold storage requirements run the risk of spoiling if their carefully temperature-controlled environments are disrupted during relabelling. Contamination is also a risk when a sterile environment is key. Tamper seals may have to be broken and the process may put randomisation in jeopardy, which is an absolutely key element to clinical drug trials. Additionally, drug sponsors will need to enable additional Qualified Person Release, further increasing costs and lead times. It’s clear to see why a lot of organisations are worried about this!
KSR: Who stands to be affected by this change?
JM: As Annex VI currently stands, organisations across the clinical trial supply chain — such as depots, CDMOs, drug sponsors and logistics firms — will feel the impact. Logistics firms and depots in particular are considering whether they now need to invest in new cold storage and cleanroom facilities to allow them to continue to serve clinical trials efforts. This will also have an impact on patients through the increased development times for new drugs as formulators adapt to the new requirements.
KSR: Can anything be done immediately to resolve these challenges?
JM: Unfortunately, there is no time left for consultation from the industry on this and the EU Clinical Trials Regulation will come into effect on 31 January whether sponsors are ready or not.
KSR: What are some solutions to these issues?
JM: To be clear, there is no “silver bullet” solution that will make it easy to achieve compliance with the new regulations. We have heard of some sponsors planning on waiting it out and hoping for a change to the regulation, but this approach lacks certainty. So, a range of solutions are required to keep potentially vital new drugs flowing through the approvals pipeline.
Technology such as e-labels could help. A digital record of required information that could be remotely updated without having to tamper or break open packs might be suitable for some applications. However, the regulation specifically demands that the label must visually show this information and so technology does not necessarily provide the answer to the challenges of Annex VI. In addition, it’s important to consider the potential costs of this, whether digital labelling technology can survive cold storage or be suitable for aseptic applications.
The entire supply chain would need to adopt e-labelling technology as well, so this does limit its short-term viability. Another issue is that the regulation specifies that physical labels must be used, so this will require an actual change to the legislation itself.
Just-in-time (JIT) labelling and investment in central distribution depots so that they are equipped to handle the relabelling and repackaging demands are other avenues being explored. JIT labelling is likely to help in circumstances when the “last-minute” labelling of drugs packaging is sufficient but can be difficult to implement and needs a reliable and predictable cadence to a trial’s progress – something that cannot be guaranteed.
Investment in depot capabilities so they can serve as a singular base of distribution with all the capabilities necessary to meet Annex VI requirements is also an option. This will be expensive and time-consuming to implement, however, and does not eliminate the risks of spoiling batches of drug through contamination or poor temperature control.

KSR: What short-term solutions can drug sponsors make use of?
JM: As labelling of the packaging is one of the key issues with Annex VI, one of the methods of overcome these challenges is through innovation in the packaging itself.
As a specialist in providing pharmaceutical packaging solutions, our team members are very much culturally aligned with the needs of clinical trial packaging. This, and our extensive record of helping drug manufacturers to develop packaging that meets their needs and regulatory obligations, gives us the confidence to say that packaging design could be the most immediate solution to Annex VI.
We have already designed packaging prototypes that allow for easy relabelling without having to break tamper seals or repackage each item through closable windows and other innovations.
The advantages to these solutions are that extensive reworks of facilities can be mostly avoided and new packaging can be adopted relatively quickly. However, we would like to again stress that there is no “one-size-fits-all” solution to this problem. Innovative packaging presents the most immediate “fix,” but adaptations across the whole clinical trials sector need to be made in the long-term.
KSR: Is there anything else readers should be aware of?
JM: As this is a European Union piece of legislation, those outside of Europe might be curious to know how this will affect them. Although we can’t speak for every non-EU pharmaceutical company, we are confident that most, if not all, will follow suit to facilitate an easier integration with customers. After all, Europe represents a sizeable portion of the catchment for global clinical trials, so we anticipate that the EU Clinical Trials Regulation will drive the standard for sponsors around the world.
KSR: What would you say in summary to this?
JM: Unintended consequences are an all-too-common occurrence when it comes to large-scale new legislation as we all know. It will be interesting to see how the EU responds to the growing complaints that drug sponsors are making as they will have to adapt to the requirements of Annex VI and this will have an impact.
It’s important to remember that the goal of the new regulation is laudable in its aim to streamline the approvals process for vital new drugs. It’s a shame, however, that such a small clause will end up causing so much disruption. We are hopeful that pressure from sponsors can help to change the requirements of Annex VI sooner rather than later.