Redesigning reactions is key to making the synthesis of drug molecules for late stage trials and commercial production cheaper, more efficient and safer. Dr Sarah Houlton looks at some recent successful examples that create better large-scale routes to chirally pure APIs.
With the importance of single enantiomer molecules in the pharmaceutical industry, it is perhaps unsurprising that the process chemistry groups within pharma companies are adept at solving chirality problems. Whether this involves applying reactions developed elsewhere or inventing their own to create better large-scale routes to chirally pure APIs, their efforts enable the practical synthesis of drug molecules for late stage trials and commercial production.
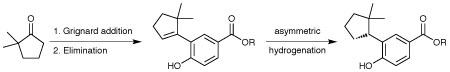
The chemical process r&d group at Amgen in Thousand Oaks, CA, US, for example, developed a catalytic asymmetric synthesis route to a tertiary benzylic carbon centre, using a phenol-directed alkene hydrogenation.1 These moieties are challenging for synthetic chemists to put together, and often functional ‘handles’ are left behind that must be removed. This is not very atom efficient. Amgen had identified a chiral phenol that was a key structural motif in the series of drug candidates, and could also be applied to the synthesis of the marketed overactive bladder treatment tolterodine (Detrol), which loses US patent protection later this year. Some form of asymmetric hydrogenation appeared to be the obvious synthetic choice.






