The delivery of therapeutic agents into cells through cell membranes (cellular uptake) is of profound importance to the industry and has always proved to be a challenge, especially in the delivery of large molecules. The past 20 years have seen a dramatic increase in the number of drugs based on larger molecules – proteins, peptides and nucleic acids – that have the potential to circumvent the limitations of small molecules. Currently there is a demand for more efficient delivery systems to administer biologicals into specific cellular targets. Issues such as poor stability in vivo, lack of cellular uptake and insufficient ability to reach targets are challenges that have been a focus for research over recent decades.
The plasma membrane acts as a barrier to the direct translocation of hydrophilic macromolecules, preventing efficient and controlled intracellular delivery. A drug must be either highly lipophilic or very small to stand a chance of cellular internalisation and it is difficult to ascertain a generic mechanism for drug uptake. These restrictions mean that the repertoire of possible drug molecules is limited. Similarly, novel therapeutic approaches, such as gene and protein therapy, also have limited potential due to the impermeable nature of cells to peptides and oligonucleotides.
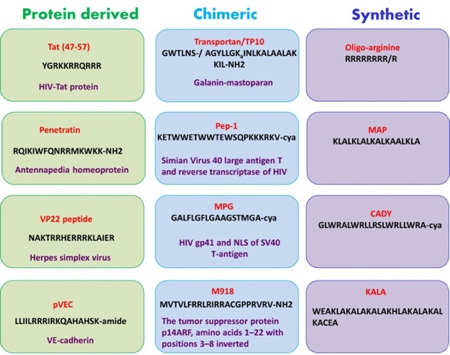
The number of known natural and synthetic peptides with cell-penetrating capabilities has continued to grow
The existing methods for delivery of macromolecules, such as viral vectors and membrane perturbation techniques, can result in high toxicity, immunogenicity and low delivery yield. A number of non-viral strategies have been introduced, such as lipid, polycationic, nanoparticle and peptide-based formulations, but only a few methods are practically used in vivo at either preclinical or clinical levels. Thus, novel efficient carrier delivery methods have to be developed to impart good bioavailability of drug molecules.






