In an ideal world, all synthetic processes would be high-yielding, selective, cheap, produce little waste and, for a chiral product, give just one enantiomer. In reality, while very many successful enantioselective synthetic reactions are available, there are some molecules for which a chirally pure process is so much more expensive and inefficient than a racemic version followed by some form of separation that it is impractical to make the single enantiomer. This is particularly the case if the racemisation is dynamic, with the ‘wrong’ enantiomer being racemised to create more of the correct one.
As a result, new methods for dynamic resolution are still being developed and used to make active ingredients and intermediates. A good example of this recently came from a group at Radboud University in Nijmegen, the Netherlands, working with industrial partners at DSM, Syncom and Agfa-Gevaert,1 using a grinding technique that was first described in 2005 by Cristobel Viedma of the geology depart-ment at Madrid’s University Complutense.2
Crystals in a slurry are ground up, which induces racemisation within the solution. It is thought the grinding splits small fragments off the larger crystals, which are more soluble than the bigger pieces. They therefore dissolve first, nurturing the larger crystals, and then subcritical clusters with the same handedness within the crystals are reincorporated into the crystals, thus giving a measurable accumulation in the solution of the other enantiomer. The overall effect is a net flux of one enantiomer into the crystals, and the other into the solution.3 Many examples have been described on small quantities of material, but the work from the Dutch groups was carried out on a much larger scale.
The Dutch group believes a bead mill would be the best way to carry out the process at scale, having already shown on smaller quantities that the intensity of abrasive grinding it produces was very efficient in turning the solids of an almost racemic mixture into its enantiomerically pure form. As an example, it looked at an intermediate on a route to the heart drug clopidogrel (Plavix), which is straightforward to racemise in solution using the organic base DBU (1,8-Diazabicyclo [5.4.0] undec-7-ene). This intermediate is a racemic conglomerate, and thus ideal to try the grinding process on.
The group looked at both small-scale and larger volume grindings as a comparison. On a small scale – 0.3g of the intermediate using 8g of glass beads, acetonitrile as solvent, and 0.15g of the racemisation catalyst DBU added after 10 minutes (min) of intense grinding in an ultrasonic cleaning bath, within six hours (hrs) the solid phase had deracemised.
A bead mill rotates at up to 4,000rpm, and with very hard yttrium-stabilised zirconium dioxide beads allows extremely intense grinding. A bead mill with 110ml free volume filled with 660g of 0.4mm diameter beads was used; the slurry was pre-prepared by dissolving 20g of racemate plus 2g of pure S-enantiomer. This was added to the mill, which already contained a further 110ml of acetonitrile to prevent it blocking. The mixture was ground at 2000rpm for 5min, and 5g of DBU added to start the racemisation in the solution phase. After 45min, the racemisation was complete – about six times faster than in the cleaning bath.
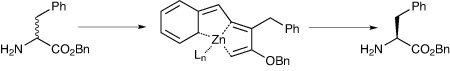
Amino acids and other chiral amines are important building blocks when constructing many APIs, but it is still common for those that do not come out of the chiral pool to be made in chiral form and then the two enantiomers separated. However, the ability to carry out a dynamic kinetic resolution is limited by the problems associated with the racemisation of the chiral amine – typical conditions for this process involve toxic metals, high loadings, and harsh reagents, and the separations are also expensive.
An alternative has been developed by Zachary Aron’s group at Indiana University, which is inspired by nature – the enzyme cofactor pyridoxal-5’phosphate, or PLP.4 This uses Schiff base to form a quinonoid and thus racemise amino acids; the conditions are mild but it does not meet the criterion of being cheap. Several analogues have been reported in dynamic kinetic resolutions, including salicylaldehyde and pyridoxal, but again, their use is limited by cost, low activity, or both.
The Indiana group has found that simple metal complexes of picolinaldehyde are able to catalyse the racemisation of amino acid esters, under conditions that still allow enzymatic resolution to take place. The idea was that the metal binding would make it easier to form the Schiff base, similar to the 3-hydroxyl group of PLP. They also thought that cation binding might improve reactivity by co-ordination to the nitrogen in the pyridine ring, which would stabilise the electron-rich quinonoid tautomer (Scheme 1).
The researchers discovered that the racemisation worked best when carried out in protic solvents, with added ammonium salts to promote the Schiff base exchange. Complexes of zinc triflate and picolinaldehyde racemised the test substrate, phenylalanine methyl ester, about four times more quickly than when the racemisation was carried out using pyridoxal, and complexes of 4-pyridinecarboxaldehyde did not work at all, presumably because of their inability to chelate the zinc.
They then tested the racemisation out in the dynamic kinetic resolution of a number of different amino acid esters using the commercial proteolytic enzyme Alcalase. This system had previously been shown to work alongside pyridoxal catalysed dynamic kinetic resolution.5
The group found it had to make some alterations to the DKR process, notably including lithium carbonate as a buffer because a protic acid is generated, and the reactions were run in a mixture of t-butanol and water to prevent the product racemising. The researchers were able to isolate highly enantioenriched products from the mixture by filtration, and good results were obtained with both straight and γ-branched chain amino acids, and also aromatic ones.
A team at Kyowa Hakko Kirin in Japan has used a crystallisation-induced dynamic resolution in the synthesis of KW-4490, a PDE-4 inhibitor being developed for asthma and chronic obstructive pulmonary disease.6 Towards the end of the synthesis, they were faced with a mixture of cis and trans diastereomers of an intermediate derived from a hydrocyanation reaction, which was about 62:38 cis:trans; altering the conditions of the reaction did not give a selective process. The desired isomer was the cis, so they wanted to convert the unwanted trans isomer to cis to improve the yield (Scheme 2).
They first tried using a base-induced isomerisation using a base such as potassium t-butoxide, but although this worked to a degree the best ratio of products obtained was 75:25. The same result was obtained when they tested the system on both pure cis and trans isomers, indicating that this ratio represented the thermodynamic equilibrium. However, they realised that the cis isomer was less soluble in ethanol, so they thought the answer might lie in crystallisation-induced dynamic resolution.
They therefore suspended a crude mixture of the two isomers in ethanol and added a catalytic amount of potassium t-butoxide to effect the isomerisation. It was stirred and warmed, and hexane added portion-wise to crash the cis isomer out of solution. The group managed to increase the ratio of isomers to 99:1 by continuous isomerisation, with a 90% isolated yield.
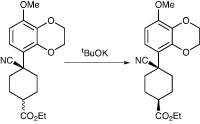
Scheme 2: Kyowa Hakko Kirin found a way to improve the yield of the cis isomer
Another way of creating a chirally pure form of a racemate is first to convert it to an achiral derivative, and then replace the chirality selectively to give just one enantiomer. This is what a team from Bristol-Myers Squibb from New Jersey did in the synthesis of an intermediate required for an antidiabetic drug candidate (Scheme 3).7 First, the racemic amino acid was deracemised enzymatically. Initially, an amino acid oxidase was used to selectively oxidise the wrong, (R), isomer to the corresponding keto acid, leaving the (S) isomer untouched. This was effected using the (R)-amino acid oxidase from T. variabilis, which had been cloned and overexpressed in E. coli, and then immobilised on Celite. The oxidation was carried out on 1.95g of racemic amino acid, and after 5.5hrs the ee had reached 89%.
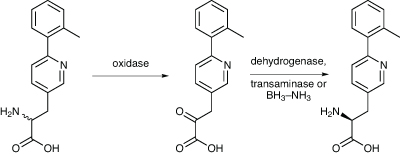
Scheme 3: Bristol-Myers Squibb managed to create a chirally pure form of a drug intermediate by enzymatic deracemisation then conversion back using a transaminase
The keto acid was then reductively aminated back to the (S)-amino acid, again enzymatically, using an S-amino acid reductase. The enzyme was chosen after a screening of microbial cultures for one that was active with a keto acid, and they identified a strain of S. ureae as producing a suitable S-amino acid reductase. So, after 7.5hrs, the S-amino acid dehydrogenase, in the form of cell extracts from S. ureae, was added to the reaction, and run for 19hrs, by which time no keto acid peak was visible on HPLC. The (S)-amino acid was isolated in 99% ee, with an overall isolated yield of 54%.
However, the two processes could not be run in the same pot as the oxidase enzyme was inhibited by the conditions needed for the reductive amination. So they tried an alternative for the second step – an (S)-transaminase instead of an (S)-amino acid dehydrogenase. A suitable transaminase was found in a Burkholdia bacterium found in a soil sample. The next step was to develop a fermentation process for the production of the two enzymes in recombinant E. coli, and this was scaled up to 250 litres, from which 11–13kg of wet cell paste from cells expressing the aminotransferase, and 7kg from the oxidase. The cells expressing the oxidase were much more active, so a 5:1 mixture of cells expressing oxidase and aminotransferase was used, giving a combined cell extract that had both activities. The reaction has been scaled up to 607g of the racemic mixture, with a 66% yield of the correct isomer being isolated, having an ee of 99.9%.
A third alternative was also explored – a chemoenzymatic dynamic resolution. The first oxidase step to produce the keto acid was still used, but this time they employed a chemical reduction of the imine that is the initial product of the oxidase reaction, using borane–ammonia complex, all in one pot. This reduces it to the racemic amino acid before the keto acid can be formed, thus enriching the correct isomer. A maximum yield of 79% of the desired single enantiomer was obtained, with ees above 99%, using 10eq of borane–ammonia complex, and stirring for 20hrs. No significant improvement in results was seen by doubling the amount of reductant used. However, they decided to go with the two enzyme process involving the oxidase and the transaminase as the process route, as it was most the efficient alternative.
The long-standing drug dexmethyl-phenidate is used to treat ADHD. It is the single enantiomer form of the older drug methyl-phenidate, and has seen generic competition for some time. The manufacturing process for an advanced intermediate involves the resolution of DL-threo-α-(2-piperidyl) acetamide using dibenzoyl-D-tartaric acid. While this process is effective, giving a good yield and an ee in excess of 99%, a large amount of unwanted L-threo-amide and uncrystallised D-threo-amide remain in the mother liquor. A team at Emcure Pharma-ceuticals in Pune, India, has developed an efficient way of racemising the wrong isomer (Scheme 4).8
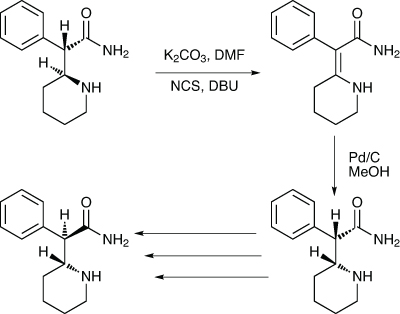
Scheme 4: Emcure has found a means to improve the yield of an intermediate in production of dexmethylphenidate
Analysis of the mother liquor when they repeated the literature procedure showed by HPLC that it contained 15–20% of the desired D-threo-amide, and 80–85% of the wrong isomer. They thought the best way to racemise the wrong isomer would be to create a double bond between the two chiral carbons by taking advantage of the reactive C-2 benzylic carbon, first halogenating it and then carrying out a dehydrohalogenation. The cheap option, they said, would be to use N-chlorosuccinimide to chlorinate it, and then DBU to effect the elimination.
The chlorination was carried out in DMF with potassium carbonate, and was complete after three hours, followed by the one-pot addition of DBU to create the double bond, avoiding the need to isolate the chloro-adduct. The optimum amount of DBU to add was 1.25eq. This was then reduced using Pd-C in methanol at 45°C, with the reaction taking 10hrs. This gave almost exclusively the DL-erythro isomer, which was then elaborated to the desired intermediate and, ultimately, dexmethylphenidate using published procedures.
Pfizer compares chemical resolution with enzymatic
Scientists at Pfizer in Sandwich, UK have developed an improved way of making the chiral intermediate (1R,3R)-3-Boc-aminocyclo hexanecarboxylic acid.9 While several syntheses of the parent free amine have already been published, the group needed a shorter route that could be run on a large scale.
The basic synthesis involves the hydrogenation of 3-aminobenzoic acid, which gives a mixture of cis and trans isomers of the cyclohexane derivative, and this is then reacted with Boc2O to add Boc protection to the nitrogen. This was then recrystallised from dichloromethane to give the cis diastereomer alone in racemic form.
Initial conditions for the hydrogenation used Raney nickel, but this gave a ratio of 3:1 of cis:trans, which improved to 5:1 when the Boc group was added. So they looked for milder and more selective conditions for the hydro-genation, and they found that rhodium on carbon gave a better ratio of 5:1 and, importantly for a production process, took place at far lower temperature and pressure. Adding the Boc group this time gave just the cis isomer, ready for resolution. This was achieved with (R)-1-phenylethylamine, using ethanol/hexane as the solvent system to avoid chloroform, which had been used before, and producing the desired compound in pure form, albeit in fairly modest yield.
They also thought it might be possible to effect the resolution enzymatically on the ethyl ester of the desired compound, giving one isomer as the free acid, and leaving the other as the ethyl ester. They ran 95 commercially available hydrolases through a high-throughput screen, and four of them appeared to carry out the hydrolysis stereoselectively. The best results were achieved with the cholesterol esterase from Candida cylindracea, which gave 99% substrate ee at 50% conversion.
references
1. W.L. Noorduin et al. Org. Proc. Res. Devt 2010, 14, 908
2. C. Viedma, Phys. Rev. Lett. 1005, 94, 065504
3. W.L. Noorduin et al. Angew. Chem. Intl. Ed. 2010, 49, 8435
4. A.E. Fellen et al. Org. Lett. 2010, 9, 1916
5. S.-T. Chen et al. J. Org. Chem. 1994, 59, 7580
6. A. Yanagisawa et al. Org. Proc. Res. Devt 2010, 14, 1182
7. Y. Chen et al. Org. Proc. Res. Devt published online ahead of print 2010, doi 10.1021/op1001534
8. A.B. Chavan et al. Org. Proc. Res. Devt 2010, 14, 1473
9. M. Badland et al. Tetrahedron Asymm. 2010, 21, 864






