An example of this is the Human Cell Atlas (HCA) project, a collaborative international effort that aims to define all human cell types in terms of distinctive molecular profiles (such as gene expression) and connect this information with classical cellular descriptions (location and morphology, for example).1
The translational promise of the cell atlas ranges from basic biology of the human organism to disease mechanism, diagnosis, prognosis, and from treatment monitoring to immunotherapy, drug development and cell and organ replacement.2
Sophisticated single-cell analysis platforms are allowing researchers to gather crucial insights from databases such as the HCA, helping them to better understand the mechanisms underlying many diseases. It is now also emerging as an important contributor to the international effort to tackle SARS-CoV-2.3
This vision of precision medicine — an ideal “new state” wherein relevant and reliable data informs clinical decisions and allows each patient to be more appropriately treated than is possible currently — is getting ever closer thanks to the technological advances being made, particularly in the field of data handling and analysis, and the groundbreaking single-cell research that this technology is enabling.
The game changer
Single-cell analysis is widely seen as a trailblazing technique; it allows omics analysis — notably genomics, transcriptomics, epigenomics and proteomics — at the single-cell level and enables, for example, the identification of minor subpopulations of cells that may play a critical role in a biological process.
It can also provide an ultrasensitive tool to clarify specific molecular responses to therapy and pathways … and thus reveal the nature of cell heterogeneity.
This extends the mantra of precision medicine from “right patient, right target” to include “right cell type.”
With this new toolbox, researchers and clinicians can look for insights into the transition from “healthy” to “disease” states, study potential biomarkers, understand the mechanics of disease pathways and assess responses to drug targets or available therapeutic regimens with time.
Cells versus patients
It is important to realise that current single-cell datasets have been generated from a small number of individuals, and statistical significance relies on the number of patients studied, rather than the number of cells.
This is because cells from the same patient are “siblings” and not true biological replicates. As a result, datasets with hundreds of thousands of patients/treatment conditions will necessitate technology to manage billions of cells.
For example, the Immune Cell Survey in the HCA currently contains 780,000 cells, but these are from only 16 individuals. The HCA itself has fewer than 400 patients, with very few donating cells from more than one organ system.
This “cells-patients-data” relationship is further compounded as researchers study the evolution of cell (sub)populations and the effects of treatments with time.
If we are to achieve data sets with the enormous population of patients and treatment conditions that are needed for statistical power in pharma R&D, the development of innovative technology to manage data from billions of cells is essential.
In the context of cell therapies, being able to monitor billions of individual cells during the manufacturing process in a cGMP-compliant manner will be crucial.

Leveraging human genetic data
Beset by inefficiency, rising costs and a challenging regulatory/reimbursement environment, Big Pharma has moved to new R&D approaches that look to leverage the vast amounts of human genetic data now being generated.4
There is a need for a single, unified repository to store an organisation’s entire single-cell data — both raw and normalised — that is organised intuitively and enables secure data transactions from unlimited users, as opposed to the persistent use of files and silos of data.
Current tools limit cross-study analysis, which is crucial to confirm results.
As a root cause, data sparsity limits the extraction of maximum value from single-cell omics data. For example, in single-cell RNA sequencing, when a gene in a given cell has no unique molecular identifiers or reads mapping to it, large numbers of observed zeros occur in the measurement values.5
The proportion of observed zeros, or degree of sparsity, can hinder downstream analysis performance and requires significant bioinformatics expertise to handle storage and computation.
Statistical models are needed that inherently account for the sparsity, sampling variation and noise modes of single-cell RNA sequencing data with an appropriate data generative model.
Case study: COVID-19
Evaluating the expression profile of key genes (such as those encoding key viral entry associated proteins) in large, single-cell datasets can facilitate testing for diagnostics, therapeutics and vaccine targets.
For example, initial work aimed at understanding SARS-CoV-2 pathology at the single-cell level — some of which used previously generated HCA data — focused on understanding which cell types expressed the gene encoding the SARS-CoV-2 receptor, ACE2.
Recent research has indicated that, in addition to the well characterised respiratory disease resulting from SARS-CoV-2 infection — the virus can cause systemic pathology including in the gastrointestinal tract, cardiovascular system, endocrine system and central nervous system.6–9
Further elucidating the relationship between these observations and the tissue/cell distribution of receptors and gene expression profiles is time-critical.
One study assessed the capabilities of a new single-cell data analysis platform (Paradigm4’s REVEAL Single Cell) in querying all cells in the HCA and COVID Cell Atlas (CCA) databases that express either the ACE2 receptor, TMPRSS2 (the entry facilitating enzyme transmembrane serine protease) or coexpress both markers (Figure 1).10
Most of the cells expressing ACE2 had a cell type tag of PC_vent1 (heart tissue); the majority of cells expressing TMPRSS2 had a cell type tag of AT2 (alveolar epithelial type II cells found in the lung parenchyma); and most cells coexpressing both ACE2 and TMPRSS2 were tagged as gallbladder cells.
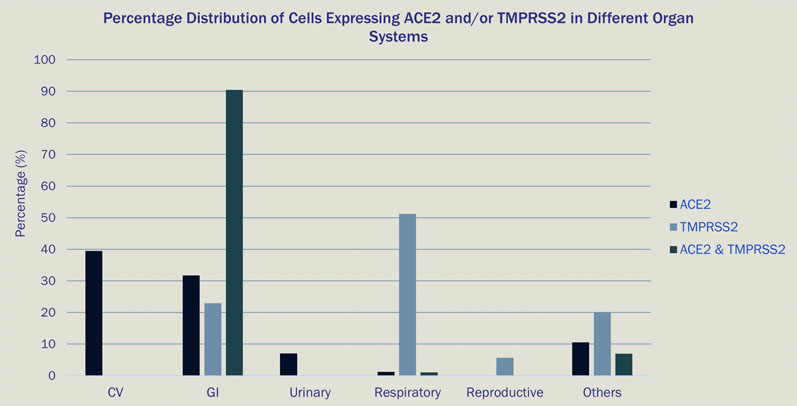
Figure 1: Percentage distribution of cells expressing ACE2 and/or TMPRSS2 in different organ systems
These results go some way to explain the multiorgan involvement in infected patients observed globally during the ongoing COVID-19 pandemic, as multiple cell types in the human body express genes utilised by SARS-CoV-2 for infection.
Paradigm4’s scientific analytics engine enables rapid (less than 60 seconds) profiling of key genes involved in the infection and supports additional use cases that require evaluation across a large database of single-cell expression datasets, such as vaccine candidates for infectious diseases, biomarkers for oncology patient stratification and immunology related disorders.
This illustrates the value of an analytics engine such as REVEAL Single Cell to power discovery across large, sparse datasets, in line with demands from the research community.
An exciting future within our reach
Single-cell analysis offers broad scope for scientists and may provide a way to uncover physiological interconnections between tissues that we had no hope of seeing previously.
How this translates into progress in precision medicine, assessing the impact of epigenetic factors and the development of predictive models for daily health, as well as therapeutic research and development, relies largely on extracting maximum value from the multiomics data being produced and assembled into cell atlases, such as the HCA.
Integrating these existing repositories with research, such as the ongoing COVID-19 efforts, would not be possible without powerful computational tools.
With optimised processing and data management and analysis methodologies to extract translational value, the ability to find gene expression fingerprints and distinct cell types that may not look like each other at all, but might be corresponding with each other, could transform the way we diagnose and treat disease.
If the full potential of single-cell analysis is realised, we will be able to navigate the physiology of humans from the molecule up.
References
- https://doi.org/10.1101/121202.
- www.humancellatlas.org/wp-content/uploads/2019/11/HCA_WhitePaper_18Oct2017-copyright.pdf.
- https://doi.org/10.1038/s41580-020-0267-3.
- www.gsk.com/media/5041/rd-update-slides-hal-barron.pdf.
- https://doi.org/10.1186/s13059-020-1926-6.
- K.S. Cheung, et al., “Gastrointestinal Manifestations of SARS-CoV-2 Infection and Virus Load in Fecal Samples from a Hong Kong Cohort: Systematic Review and Meta-Analysis,” Gastroenterology 159(1), 81–95 (2020).
- https://doi.org/10.1101/2020.04.06.028522.
- N.P. Somasundaram, et al., “The Impact of SARS-Cov-2 Virus Infection on the Endocrine System,” Journal of the Endocrine Society 4(8), bvaa082 (2020).
- S. Najjar, et al., “Central Nervous System Complications Associated with SARS-CoV-2 Infection: Integrative Concepts of Pathophysiology and Case Reports,” J. Neuroinflammation 17, 231 (2020).
- https://doi.org/10.1101/2020.06.24.169730.