According to national and international laws, animal use in research should follow the 3R principle of reduction, replacement and refinement. When in vitro and, to some extent, in silico systems can provide the information needed, they should be prioritised instead of animal studies. When animals are inevitable (as is the case in vaccine toxicity investigations), the following approaches are recommended to be adopted.
Literature review and use of available data: study type and design should be customised to meet specific scientific objectives to generate maximum data with the minimum number of animals (use of default study protocols/standard templates are discouraged).
A detailed review of all available toxicology/safety data for the vaccine components and learnings from similar vaccine types will aid in identifying the potential issues and data gaps with the candidate vaccine. At times, this may help to come up with a study design that involves a smaller number of groups with fewer endpoints or a shorter duration of administration that will still answer the key safety questions (Tables III and IV).
Frontloading of toxicity evaluations: the initial non-clinical studies are aimed at determining the activity of the candidate vaccine in relevant animal models. In addition to evaluating immune activation/potency, certain safety endpoints can also be incorporated without interfering with the primary objective of the study.
For example, in-life observation of animals and the measurement of body weight, feed consumption, haematology and clinical chemistry can easily be included as a measure of adverse effect. At sacrifice, critical items, including immune organs, can be collected and, if needed, microscopy done to identify any adverse changes even prior to initiating a formal toxicity study.

Sebastian Joseph
Integration of multiple endpoints into toxicology study: to save time, money and animal use, it is recommended to include multiple endpoints in a given toxicity study. For example, safety pharmacology endpoints, the potential to impair male or female fertility, pharmacodynamic characterisation, etc., can be included as endpoints in a repeated dose toxicity study.
GLP versus non-GLP
All pivotal toxicology studies should be done in compliance with the principles of good laboratory practice (GLP), such as 21 CFR Part 58, OECD’s principles of GLP or other appropriate standards. Certain immunological endpoints, which are part of these pivotal GLP studies, may be unconventional and might have to be evaluated in a lab that does not operate in compliance with formal GLP.
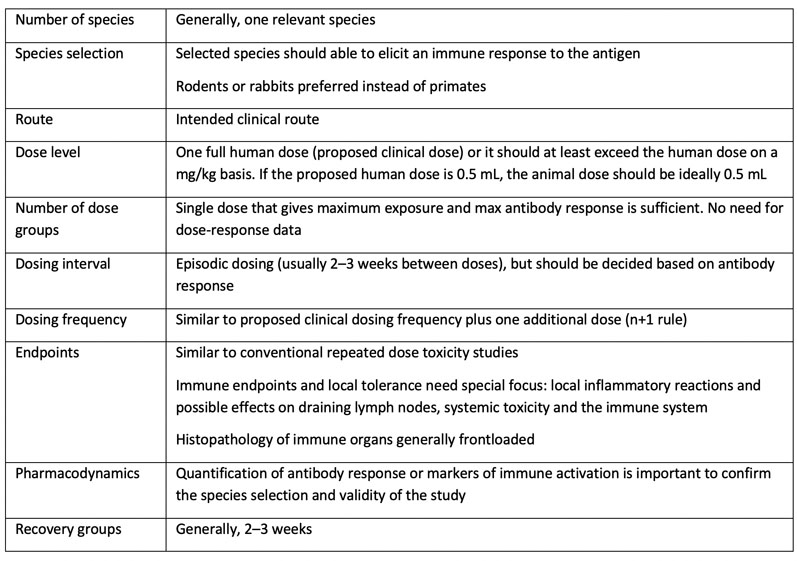
Table III: A typical study design for a general toxicity study of a candidate vaccine
Another scenario is that safety information may come from pharmacology studies that are not typically done in compliance with GLP. In these two scenarios, the lack of GLP compliance does not disqualify the data — provided the studies were well conducted and documented, and are available for review if requested by an agency.
Can non-clinical safety studies be bypassed to fast-track clinical development?
In general, non-clinical studies should support the proposed clinical trial design. For vaccines, the pivotal safety investigations are done prior to first-in-human (FIH) trials, especially if the trial designs comprises repeat administrations. When there is an urgency to develop a vaccine owing to an ongoing medical crisis, as in the case of COVID-19, certain provisions to relax the non-clinical study requirements may be possible.
For example, in response to the COVID-19 pandemic, in March 2020 the US FDA allowed Moderna Therapeutics to FIH test its mRNA-based candidate vaccine, mRNA-1273 (within a total of 63 days from sequence selection to first human dosing).7
In June 2020, FDA issued guidelines in which it allowed a human trial to be initiated without non-clinical toxicity studies in situations when adequate information to characterise product safety may be available from other sources.8
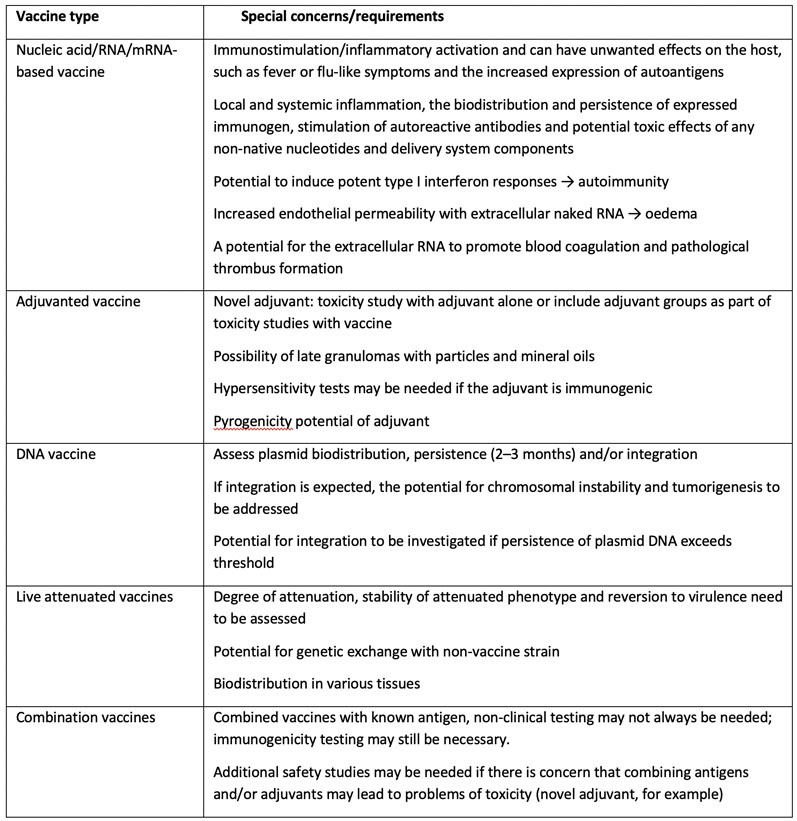
Table IV: Special safety concerns associated with different vaccine types
For example, if the COVID-19 vaccine candidate is made using a platform technology utilised to manufacture a licensed vaccine or other previously studied investigational vaccines and is sufficiently characterised, it may be possible to use toxicology data (data from repeat dose toxicity studies, biodistribution studies, for example) and clinical data accrued with other products using the same platform to support FIH clinical trials for that COVID-19 vaccine candidate.
In summary, non-clinical programmes should be customised to accommodate various aspects of the investigational vaccine. In general, non-clinical safety studies are abridged when compared with non-vaccine pharmaceuticals; however, novel additives will require extensive investigations.
Among all the effects, any potential adverse effects on the immune system and local tolerance are the focus areas of the non-clinical safety programme. The possibility of adopting lean study design should be explored to save time, money and animal usage. As the issues with each vaccine can vary, early dialogue with the relevant agency will help to avoid the need for additional studies during the latter part of clinical development or prior to approval.
References
7. www.modernatx.com/modernas-work-potential-vaccine-against-covid-19.
8. www.fda.gov/media/139638/download.
Part I can be read here.






