
The CMC challenges in in vivo CGT are significant because of the complexity of the platform and predominantly revolve around ensuring the safety, efficacy and consistent quality of the products.
For viral delivery systems, these challenges include optimising viral vector production to ensure adequate gene delivery, developing robust purification techniques to eliminate impurities and ensure product safety, and establishing reliable and standardised manufacturing processes to meet regulatory requirements.
For non-viral vector systems, such as lipid nanoparticle (LNP)-based gene therapy, these challenges include establishing consistent and scalable manufacturing processes to produce the LNPs with precise size and structural characteristics, ensuring the stability and integrity of the genetic material during encapsulation and storage, and developing robust quality control measures to assess the purity, potency and safety of the final products.

For gene editing technologies, issues include the development of precise and efficient delivery systems to target specific cells or tissues, ensuring the accurate and controlled editing of the target genes to avoid off-target effects and potential genomic instability, and establishing standardised and reproducible manufacturing processes to produce CRISPR-Cas9 components.
Implementing rigorous quality control measures to monitor the integrity and activity of guide RNAs and Cas9 proteins throughout the manufacturing process is essential to ensure the safety, efficacy and consistency of gene editing therapies.
Additionally, ensuring long-term stability, scalability and reproducibility of the gene therapy product presents further challenges.
Mitigation strategies to overcome CMC hurdles
Phase-appropriate sourcing for raw materials, notably plasmids, in manufacturing is crucial as they carry the genetic payload of the final drug product. Requirements for time, cost, quantity and quality differ depending on the product’s developmental stage.

Although research-grade materials suffice in early stages, scaling up to high-quality ones that conform to good manufacturing practice (GMP) principles becomes necessary for later clinical phases. In the context of CGT, early GMP implementation is crucial as pivotal clinical trials occur earlier than for traditional small molecules.
The quality of the Master Cell Banks and the production of packaging plasmids, such as lentiviral vectors used in chimeric antigen receptor (CAR) T cells and other CGT products, also significantly impact quality.
For three of the four plasmids required, namely the envelope and helper plasmids, the use of universal off-the-shelf options reduces time, cost and risks, allowing developers to concentrate on the gene of interest.
Many contract development and manufacturing organisations offer universal plasmid testing using the same methods for “research” or “GMP” materials. Although they use more lenient acceptance criteria for research-grade material, a Certificate of analysis (COA) is still required for downstream production.
Starting and ancillary materials: Given the inherent variability and complexity of starting raw and ancillary materials, establishing material qualification and management protocols is crucial.
It is also helpful to understand the critical material attributes to better control the quality of incoming material. For autologous products where there is limited availability of starting materials, identifying independent backup suppliers for critical raw material is recommended as is keeping an additional supply of the material in house.
For allogeneic products, selecting and establishing a quality agreement with the supplier for higher donor eligibility determination is recommended.
Manufacturing process: CGT products have a relatively high risk of contamination owing to the multiple sites and interventions involved. When possible, apply a semi- or fully automated process as well as single-use disposable processing components.
Adopting a closed system could also minimise handling steps and subsequently reduce the chances of contamination.
Technology transfer likely occurs during the product development phase. Therefore, it is a good practice to establish a well-defined tech transfer strategy to address both timeline and resources at the transferring and receiving sites.
Tech transfer should be performed when the critical quality attributes (CQAs) of the final products and in-process parameters are well understood. Establishing the transfer acceptance criteria as a measure of success for the tech transfer is also recommended.
Process changes are inevitable in CGT product development; therefore, the sponsor needs to plan, monitor and evaluate these changes and provide data to support them.
It is recommended that sufficient samples are retained to prepare for future comparability studies when process changes occur. Sponsors are also encouraged to identify key and critical process parameters (CPPs) as early as possible to establish a control strategy.
Supply chain: As supply chain complexity remains a CMC challenge, proper handling including storage, shipping conditions, chain of identity and chain of custody needs to be considered throughout the process — from the collection of the starting material until the final product has been administered to patients (Figure 1).
To reduce variability in starting material, it is recommended that the sponsor establishes an apheresis collection standard operating procedure, which includes training the healthcare personnel who will be performing the collection procedure to ensure that the process is consistent across the clinical sites.
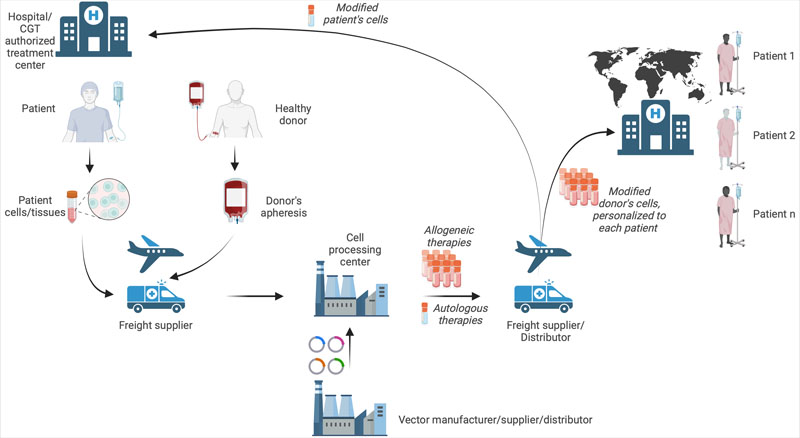
Figure 1: The CGT supply chain
Process/quality control: The analytical procedures involved in characterisation and release testing for CGT products, such as potency assay and replication competent virus testing, are also more complicated.
For autologous products, the constrained sample size and absence of reference standards hinder analytical procedure development, thereby limiting product characterisation and hampering the establishment of specifications.
This challenge can be alleviated by implementing rapid testing methods for timely product release and utilising advanced technologies such as next-generation sequencing and multiplex degenerate polymerase chain reaction.
The product specification is a dynamic document that can be continuously refined, with acceptance criteria ideally based on both healthy donor and patient materials to mitigate potential deviations owing to out-of-specification situations.
For potency assay development, the mechanism of action (MoA) is often unknown or poorly understood.
This situation is further complicated by limited knowledge of the clinical response, lack of suitable animal models and a relatively short timeframe in which to manufacture and administer the product to patients.
Sponsors are advised to explore multiple product attributes using an orthogonal approach; for example, analytical development for CAR-T therapies would benefit from tumour killing, segregation of cytokines and cell marker phenotyping assays.
Part II will shine a spotlight on issues such as stability/shelf life, comparability studies and the current regulatory environment.




