It becomes progressively difficult for bile to flow through them, causing it to back up and damage the liver. Other common symptoms include itching.
Cirrhosis results as scar tissue gradually replaces healthy cells and, ultimately, liver failure results if its progression cannot be slowed by treatment with the bile salts ursodeoxycholic acid (UDCA) or obeticholic acid.
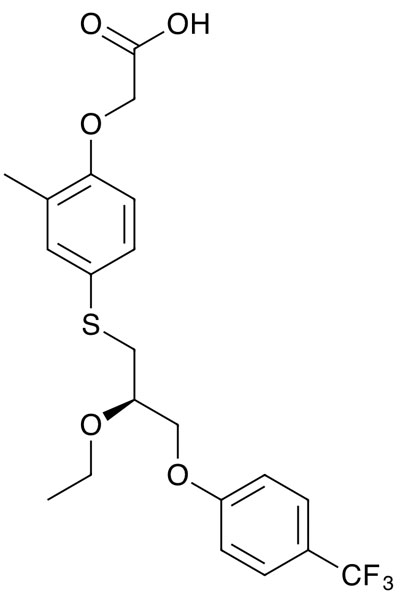
If it reaches the liver failure stage, the only remedy is a transplant. An alternative treatment, seladelpar, is a peroxisome proliferator-activated receptor delta (PPAR-delta) inhibitor that was originally developed to treat non-alcoholic steatohepatitis (NASH) by CymaBay Therapeutics (now acquired by Gilead).
It suppresses the synthesis of bile acid by reducing hepatocyte CYP7a1 activity via the FGF-21 pathway. In an open label one-year Phase II study, patients with the disease who did not have an adequate response to UDCA were given daily oral doses of 5 or 10 mg of seladelpar.1
Patients could then enrol in an open-label long-term study. The change in Globe score — a validated risk assessment tool that estimates transplant-free survival — after 2 years of treatment was assessed.
Of the 101 patients who entered the long-term study, the drop in Globe score was 0.330 after 3 months, 0.334 after a year and 0.417 after 2 years.
In a Phase II randomised, dose-ranging open-label study, 119 patients with primary biliary cholangitis at risk of disease progression — who were either receiving or intolerant to UDCA — were given 2, 5 or 10 mg of seladelpar daily for 12 weeks.2
Treatment could then be continued at 10 mg. The primary endpoint was the change in alkaline phosphatase levels from baseline to week 8.
The reductions were 26%, 33% and 41%, respectively, with responses maintained or improved after 52 weeks following dose escalation.
Pruritus scores fell in the 5 mg and 10 mg groups. There were no treatment-related serious adverse events.
A double-blind, placebo-controlled Phase III trial has also been done.3 In all, 193 patients who either had an inadequate response to UDCA or unacceptable side-effects from it were given 10 mg daily oral doses or a placebo.
Most also received UDCA as standard of care background therapy. Of those given seladelpar, 61.7% had a biochemical response, compared with 20.0% with the placebo, and 25% had normalised alkaline phosphatase levels.
By contrast, none in the placebo group did. There was also a greater reduction from baseline in pruritus scores with seladelpar. Adverse events occurred at a similar rate throughout the two groups.
References
- B. Hansen, et al., EASL International Liver Conference 2022 (22–26 June, London, UK), Abstr. Thu461.
- C.L. Bowlus, et al., J. Hepatol. 77, 353 (2022).
- G.M. Hirschfield, et al., N. Engl. J. Med. 390, 783 (2024).




