The faster they can be screened, the quicker the lead optimisation process will be, and the sooner it can undergo testing for activity. But once a molecule moves into preclinical and then clinical development, much larger quantities are required, and the synthetic route used on the lab bench is rarely appropriate.
Process chemists then have to redesign the synthesis, avoiding expensive reagents and separations as far as possible, reducing the number of steps and using more atom-efficient reactions, and ensuring that the purity of the active pharmaceutical ingredient (API) will be acceptable to the regulators.
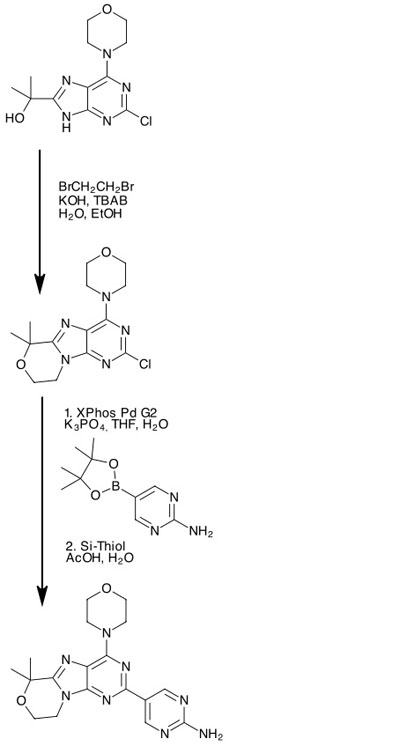
A significant reduction in process mass was achieved by chemists at Genentech in their re-engineering of the synthesis of GDC-0084, a selective dual PI3K/mTor inhibitor that is being investigated as a potential treatment for brain cancers.1
The drawbacks of the discovery route included multiple expensive chromatographic separations and the presence of trace heavy metal impurities, but they also identified that two steps could be eliminated by replacing separate addition and cyclisation steps with a direct annulation to form the structure’s fused morpholine moiety (Figure 1).
Although the first version of the new route produced nearly 4kg of the API, the annulation also generated a large quantity of side-product with a pendant morpholine. This, the desire to identify a more efficient catalyst for the subsequent cross-coupling, and the requirement to minimise the formation of a problematic acetamide impurity, led to further optimisation of the route.
They alighted on the use of a phase transfer catalyst in the annulation reaction. Adding tri-n-butylammonium bromide allowed the reaction to proceed in water without the need for organic solvents, and reducing the temperature gave fewer impurities. The product could be directly crystallised out of the reaction mixture.
For the Suzuki cross-coupling reaction, the biggest problem was the high catalyst loading at 2 mol%, allied to a very high solvent volume. A catalyst screen showed that Buchwald’s second generation XPhos PD G2 catalyst was more active than the PdCl2(dppf) that was used in the original synthesis; the lowest catalyst loading that still allowed the reaction to go to completion was 0.5%. The product precipitated out of the crude reaction mixture on the addition of water.
Finally, the palladium scavenging purification purification was optimised to prevent the formation of an acetamide impurity that was previously present at levels close to the maximum permitted. This was achieved by reducing the acetic acid: water ratio, and as the palladium loading had been reduced in the previous step, a much smaller amount of Si-thiol was required to reduce the residual quantity of palladium below the permitted 10ppm.
The improved route was used to make 6.4kg of the API, with numerous inefficient processes such as solvent exchanges and extractions having been eliminated, and the overall total solvent volume reduced by two thirds.






