Some health conditions still have no treatments while the drugs invented to treat others are less effective than they might be. Dr Sarah Houlton describes the recent efforts of medicinal chemists to address these challenges
If it were not for the skills of medicinal chemists, most of the drugs that have revolutionised the treatment of many different diseases and conditions in recent decades simply would not exist. By designing molecules that have the desired activity – and none of the unwanted activities that cause side-effects – chemists have ensured that the modern drug arsenal contains many effective medicines.
However, while many of the ‘easy’ targets are now well served, numerous diseases remain where drug therapies are less effective than they might be, and some are still untreatable. So many challenges remain for medicinal chemists. Numerous recent drug discovery projects were described at the recent International Symposium on Medicinal Chemistry, and these are some of the highlights.
Modern drug cocktails are very effective at keeping HIV at bay, but because of the tendency of the virus to mutate, it becomes resistant to existing drugs over time. Pfizer’s Tony Wood explained how medicinal chemists designed a non-nucleoside reverse transcriptase inhibitor, or NNRTI, with the express intention of avoiding resistance. They analysed the binding sites of the first generation NNRTIs, and determined that a successful new molecule was likely to be small and use hydrophobic binding as its predominant means of interacting with the receptor, and any hydrogen bonding would be to the main protein chain of the receptor, rather than to a side-chain.
One of the existing NNRTIs, capravirine, looked particularly interesting, he said. Although its pharmacokinetics were very poor, more than two mutations were needed to generate viral resistance, so there was something about the way it was using the virus’s binding pocket that was advantageous. Several elements of its structure looked like obvious causes of its negative properties – it had a pyridylmethyl group that was likely to be the source of its P450 inhibition problems, and the thioether function would render it susceptible to oxidation.
The first step was to remove the 4-pyridyl group, and swap out the oxidation-vulnerable thioether methylene group; additionally, for patentability reasons, the imidazole core was switched to a pyrazole. ‘This got us to our first active compound, which while it’s not quite as potent as capravirine, it’s much smaller as the big pyridyl group has been taken off, but it retains a reasonable degree of potency,’ he said. ‘More importantly, it’s starting to look a lot more stable, and there were no P450 interactions.’
Next, they investigated the substitution pattern around the pyrazole to optimise hydrophobic binding. ‘It’s often a good tactic to switch methylene groups around the molecule, and going from capravirine’s isopropyl/methyl to two ethyl groups gave a slightly more potent compound that was also symmetrical, which made synthesis easier,’ he explained. ‘But we still had relatively poor stability, so the next obvious step was to switch out the benzyl methylene, as it is still the most vulnerable site to oxidation, and we replaced it with a biaryl ether. This increased the oxidative stability of the compound in a human liver microsome screen from 18 minutes up to 90 minutes.’
There was still relatively high clearance as the glucuronidation-sensitive alcohol remained, and the key to fixing this was to lower its log D, which should reduce unbound clearance. They realised that they could switch the chlorine for a nitrile group – a common medicinal chemistry tactic – and this retained a reasonable proportion of the antiviral activity, but unbound clearance was greatly improved. Finally, they looked at optimising the substitution around the aryl ring, and after screening several differently substituted analogues, the di-cyano substituted compound was chosen.
‘Although it isn’t the most potent compound we made, it’s by far the most stable, with the best clearance in hepatocytes, and the lowest log D,’ Woods said. This molecule is now known as lersivirine and, unusually, it is actually somewhat smaller than the initial lead molecule – typically, medicinal chemistry programmes create larger molecules in the search for increased affinity. It is now in Phase IIb clinical trials at ViiV Healthcare, the HIV joint-venture company set up a couple of years ago by Pfizer and GlaxoSmithKline.
Diabetes therapy
Type II diabetes is a major disease burden globally, and many different strategies for drug treatment are being investigated. One target that has received a lot of attention is sodium-glucose transporter protein. There are two forms – SGLT1 and SGLT2; most glucose resorption results from the activity of SGLT2, so a selective inhibitor of this protein could provide a new form of drug therapy for diabetes that might be able to reduce glucose levels independent of insulin, as it targets the kidneys. It would need to be selective because of the additional biological activity of SGLT1, which could lead to glucose/galactose malabsorption.
One company pursuing this strategy is Bristol-Myers Squibb, as William Washburn explained. The starting point was the naturally occurring compound phlorizin, found in the bark of several fruit trees, which has renal glucose transporting activity. But it is not particularly selective, and requires large doses, as it is very susceptible to hydrolysis.
Ideally, a drug would have once daily dosing, be metabolically stable to glucosidases – unlike phlorizin – and be selective for and potent at SGLT2. A compound from Wyeth did appear to be active in mice but not rats or cell-based assays, and they postulated that in vivo glucosylation was required to activate it. This proved to be the case – it is glucuronated rather than glucosylated in the rat. However, it was still susceptible to glucosidases, and all the obvious tricks caused it to lose activity, forcing them down the prodrug route.
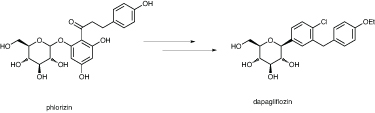
Bristol-Myers Squibb converted phlorizin to dapagliflozin
At about the same time as the BMS chemists were getting their teeth into the project, Tanabe published a patent that showed it was thinking along the same lines. It was also looking at glucosylated analogues, and the selectivity had been trebled to about 30:1, but the O-glucosylation still posed problems. One tiny by-product that was seen in a synthesis had C-glucosylation instead, and working with these types of compounds instead proved a breakthrough.
They found meta-C-aryl glucosides were more potent than either ortho or para ones, and including a methylene spacer between the rings is even better. Small lipophilic para substituents increase potency against SGLT2, and this initial series gave more than 100 times better selectivity over SGLT1.
Ultimately, the compound now called dapagliflozin was identified; this has chlorine and ethoxy substituents on the aromatic rings. It is a reversible, competitive inhibitor of SGLT2, with no off-target interactions. The molecule had a dramatic effect in obese rats, but makes them hungry – the weight loss is modulated by an increase in caloric consumption. Many other SGLT2 inhibitors are now being developed by at least eight other companies, and dapagliflozin is now in Phase III trials, being co-developed with AstraZeneca. Doses of 10mg a day have thus far proved safe, and reliably decrease glucose levels.
A different approach to diabetes, investigated by scientists at AstraZeneca, was described by Craig Johnstone. The enzyme glucokinase, expressed in the liver and pancreas, controls the rate of glucose phosphorylation and thus utilisation in the liver, and also glucose-sensitive release of insulin in the pancreas. Activating the enzyme should reduce glucose levels: individuals with a mutation that inactivates it develop a rare form of diabetes, and if they have a rare activating mutation, then hypoglycaemia and hyperinsulinaemia result.
A promising hit was found through a high-throughput screening programme, but this had a number of undesirable features, including a styrene, and an aromatic sulfur substituent. The styrene could be replaced with an amide linkage, which was still reasonably active in both mice and rats, but the unbound clearance remained too high. This was reduced by introducing a stereocentre in a sidechain, and stripping back the ether group to a smaller linking group was even better.
Both of these last two molecules were progressed, but exhibited testicular toxicity in safety studies. They speculated this could have been caused by the metabolic release of methoxyethanol, which is metabolised to methoxyacetic acid, a known testicular toxin, and the structure was modified to reduce the production of toxic metabolites. They found that when the acid function was replaced with a pyrazole, the good properties were retained, if not improved; there was good exposure and in vivo activity, with no testicular toxicity. This was the key to producing the clinical candidate AZD1092, which was well tolerated in preclinical safety studies in both rats and dogs, although it was later discontinued.
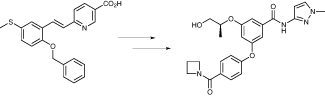
AstraZeneca remodels a promising molecule in its quest for a diabetes treatment
Fasting plasma glucose levels are closely related to hepatic glucose production, and so if it’s possible to control hepatic glucose output, it might be possible to treat patients in the earlier phases of diabetes, as well as later on. Gluconeogenesis is elevated in Type II diabetes patients, and the enzyme fructose 1,6-bisphosphatase is highly regulated by nature as a control point of the gluconeogenesis pathway. ‘People with a genetic deficiency of this enzyme are normal if they do not undergo prolonged fasting,’ said Merck’s Qun Dang. ‘So it should be safe.’
The starting point was adenosine monophosphate, which, unsurprisingly, has poor drug-like properties, and so a structure-based drug design programme was initiated. ‘We could cut out the ribose linker, replacing it with furan, and get novel purine-type heterocycles that would be more stable,’ he said. This proved equipotent with AMP, but potency could not be increased further.
Switching the scaffold to a fluoro-benzimidazole did the trick, and improved potency more than 10-fold; however, it still had low oral availability because of the charged phosphonic acid group. Even making prodrugs of this did not work as they had low oral bioavailability, and the scaffold was too big, with a molecular weight in excess of 600. A major scaffold redesign to make it smaller bore fruit – not only did a thiazole alternative prove more active, it was much more druglike.
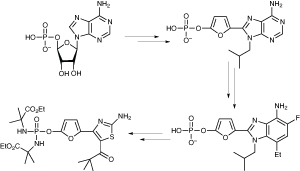
Merck converts adenosine monophosphate
The remaining problem was N-acetylation in vivo; this was a challenge as the 2-amino group is essential for binding activity, and cannot be either replaced or substituted. The solution lay in exploring the steric and electronic effects at the remote C5-position, where incorporating a bulky keto group not only prevents N-acetylation, but also maintains potency. This compound, MB07903, is now in clinical trials; it has a greatly improved metabolic profile, with good oral bioavailability, a longer half-life and reduced PK variability. In a Phase IIa trial, it significantly lowered blood glucose in diabetic patients.
There are several forms of pulmonary hypertension. Treatment options are limited for some, while others remain essentially untreatable. Nitric oxide activates soluble guanylate cyclase (sGC), which is involved in the pathogenesis of many cardiovascular diseases, including pulmonary hypertension. Stimulating sGC might help, as it should amplify nitric oxide levels, as Bayer Schering Pharma’s Joachem Mittendorf explained.
The starting point was a high-throughput screen of the company’s library, which gave a furane hydrazone lead. As well as stimulating sGC, this inhibited platelet aggregation via cGMP, and relaxed preconstricted rabbit aorta. However, its potency was not good, so a medicinal chemistry programme was instituted.
First, the benzyl group was altered, and introducing an ortho-fluoro substituent improved potency. Next, they looked at altering the indazole core, introducing a further nitrogen atom into the six-membered ring, which also improved potency; they found the nitrogen in the 2 position was essential as indoles lost all activity, as did 6,6-bicyclic alternatives.
A major breakthrough came when they modified the southern part of the molecule. Switching the furan group for an aminopyrimidine gave an order of magnitude improvement in activity, and with a cyclopropyl substituent the lead compound was 100-fold more potent than the initial hit. Its interaction with sGC is highly specific, and it is orally active, with no development of tolerance, unlike nitroglycerine, where tolerance slowly builds up and stops it from working. However, its metabolism and pharmaco-kinetic properties were not good – it has low metabolic stability and high clearance, and plasma concentrations are non-linear. It also inhibits P450 enzymes. To try and improve this, they played with the substitution patterns on the pyrimidine. Small substituents made the P450 problem worse, but a diaminomorpholine group proved very potent. It had no P450 issues, but high clearance and low oral availability.
Altering the substitution pattern around this ring provided the solution, in the form of a methyl carbamate ester. This compound, riociguat, is now in Phase II trials for two forms of the disease – pulmonary hypertension and chronic thromboembolic pulmonary hyper-tension. ‘It has a half-life of 5–8 hours, and so is administered three times a day,’ Mittendorf said. ‘This is no problem in this indication as the disease is so devastating.’
When developing small molecule drugs, the aim is usually to make compounds that are small and well absorbed as they act systemically. Sanofi-aventis’ Heiner Glombik described a project where the aim was the precise opposite – to discover a molecule for lowering cholesterol that acts non-systemically, preventing the absorption of cholesterol in the gut. ‘Non-systemic targets have the inherent advantage of avoiding many side-effect and toxicity issues, if they can be modulated by non-systemic drugs,’ he said. ‘This prerequisite is fulfilled by molecular targets located on the luminal side in the GI tract.’
Their starting point was the existing cholesterol absorption inhibitor ezetimibe, which is efficiently absorbed; the active form is made by in vivo glucuronidation, and this form passes through the liver and bile to the active site, the transport protein NPC1L1, in the small intestine. The idea was to modify this molecule so that it is not absorbed, and goes straight to the active site without any need for activation; they were looking for an analogue with a molecular weight greater than 500, plenty of potential for hydrogen bonding, and a high polar surface area.
All three phenyl rings on ezetimibe could be substituted, and they created about 100 analogues to test. One idea was to make DABCO derivatives, both by direct substitution and through spacers; another was to add fatty acid amines or taurine derivatives with a 12 carbon spacer. This last strategy, when added to the SE ring, gave good activity but was quite efficiently absorbed.
A polyol substituent with a 12 carbon spacer attached to the SE ring by an amide linkage was both poorly absorbed and active in a screen in fasted mice; this is canosimibe, the candidate drug. It has a molecular weight of 810, with eight hydrogen bond donors, 13 H-bond acceptors and high lipophilicity, as well as having low absorption in an in vivo rat model. It has a slow transit time through the gut, and minimal amounts reach the liver.
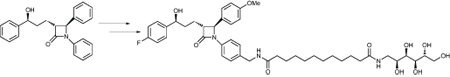
Sanofi-aventis increased the size of the compound to slow transit through the gut
In Phase II trials, it showed a significant reduction in LDL cholesterol, but it did not demonstrate the desired efficacy in a Phase III study, and it was discontinued. However, it remains an interesting example of where the traditional aims of a medicinal chemistry programme were turned upside down to ensure poor drug absorption.






