Gerardo Gomez González, Director, Service Line Leader for QMC-US and CQV SME at PharmaLex, encourages organisations to follow a dynamic validation model that can support the efficient and optimal operation of their commissioning, qualification and validation (CQV) programme.
Other related terms such as building commissioning (Cx), C&Q (commissioning and qualification), Q&V (qualification and validation) and process validation suggest additional or submodels within the larger proposed CQV operational model.
However, experience shows that they are often loosely defined and even used interchangeably.
The use, advantages and drawbacks of these models and their applicability to each of the five elements subjected to validation — facilities, utilities, systems, equipment and processes — need to be understood to determine the right validation model in any given situation.
The wider “CQV Operational Lifecycle Model” is best understood within the context of regulatory expectations and best industry practices.
The following analysis explores well-known validation models, how they fit into a CQV programme and observed best practices to support and enhance these models.
Validation models
The most common validation models in the biopharmaceutical industry are
- the V-model3
- the W-model4
- risk-based models for C&Q: ASTM-E2500 (2007) and ISPE’s Science & Risk-based C&Q Model (2019)5,6
- FDA’s Process Validation Model (2011).1
The V-model
Since its appearance in 1994 as a model for system (software) development lifecycle (SDLC) and its subsequent adaptation for equipment qualification in 2001, the V-model continues to be a constant reference in validation literature and across many CQV programmes (Figure 1).3,6
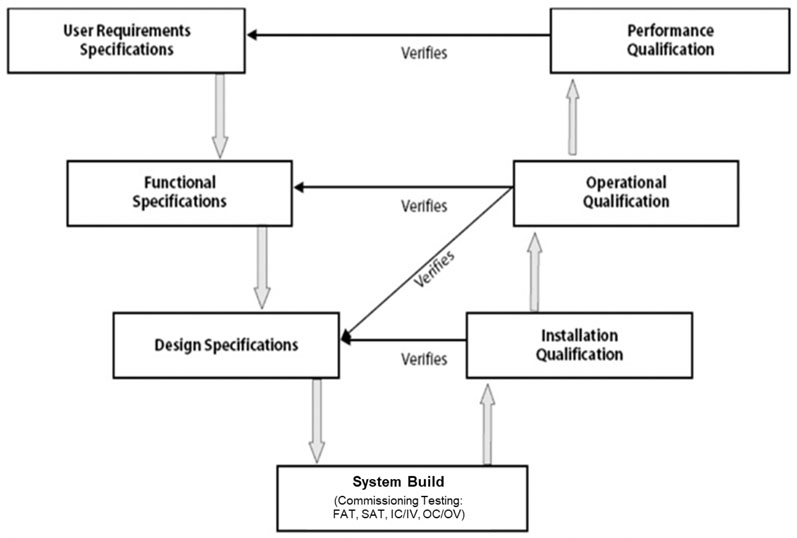
Figure 1: Visualisation of the validation V-model3
One side of the V sets the specifications whereas the other side refers to the testing conducted against those specifications to formally qualify the equipment or system.
The bottom of the V is where the equipment or system is built. Implicitly embedded in this model is commissioning testing, which has traditionally been done in the form of factory acceptance tests (FATs) and site acceptance tests (SATs).3
Depending on the system and the involvement from the vendor, these standard tests may be supplemented — or even replaced — by additional on-site testing.
These tests include installation commissioning (IC) or installation verification (IV) followed by operational commissioning (OC) or operational verification (OV).
The largest criticism of this model has been its lack of reference for science and risk-based elements.5,6 Specifically, the V-model is silent about product and process understanding, which constitute the science that would feed into — and otherwise enhance — the specification side of the V.
The options to potentially simplify the testing side of the V in the form of risk assessments are also not shown in this model. In other words, the links with ICH Q9 and ICH Q8 are not illustrated.7,8
Nonetheless, for a representation of the fundamental CQV requirements, the V-model remains valid.
The W-model
Although not set up as a risk-based model, analysis indicates that the W-model (Figure 2) paved the way for subsequent iterations that use quality risk management (QRM) to simplify qualification efforts by bringing the commissioning elements outside of the “System Build” box in the V-Model.
The “C elements” are explicitly represented as a centre portion.4
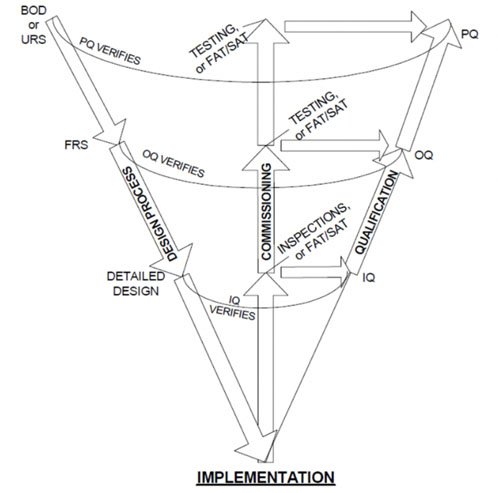
Figure 2: Visualisation of the W-model4
In this analysis, the W-Model added more granularity to what was later identified as “verification testing.” That is, both commissioning testing (such as FATs and SATs) and qualification testing (IQ, OQ, PQ).
Risk-based models for C&Q
Between 2004 and 2009, the concept of QRM was highly publicised and promoted by the US Food and Drug Administration (FDA) and the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH Q9).7,9
This, together with increased awareness about product and process understanding, also promoted by ICH (ICH Q8 ), led to the first official risk-based validation model.8
It was introduced as a standard in 2007 by the American Society for Testing and Materials and identified as “The Specification, Design and Verification Process.”5 See Figure 3.
During discussions about this model, there have been some observations that it involves less granularity of testing: calling it “verification” and removing all references to both commissioning or qualification.
In general, however, the benefits far outweigh any of its potential drawbacks. It allows for more agile navigation through the validation journey by introducing Risk Management and by emphasising the presence and involvement of subject matter experts (SMEs) with product and process knowledge — balanced with adherence to regulatory requirements and the company’s own quality procedures.5
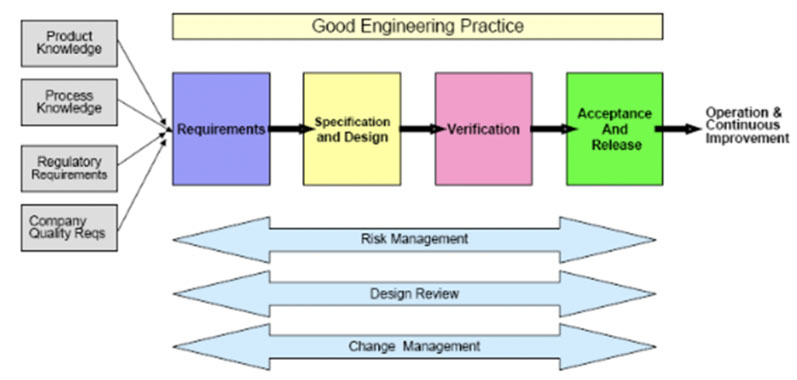
Figure 3: The specification, design and verification process from ASTM E25005
As such, this model supports leveraging vendor qualification packages and applying traditional commissioning testing, such as FAT and SAT tests, to the qualification phase.
In 2019, the International Society for Pharmaceutical Engineering (ISPE) published a revised version of its Baseline Guide for Commissioning and Qualification.6
The model discussed in this guideline replaces the V-model with an integrated C&Q model that relies on having SMEs from the engineering and equipment side work alongside other experts from regulatory and quality to establish the user requirements.
Once the initial requirements are established, each facility, utility, system and machine is classified as having either a direct or non-direct impact on product quality.
The model calls for an improved QRM process that feeds into a Design Review/Design Qualification process iteratively. More granularity is provided by the 2019 ISPE Baseline Guide as to the type and extent of the verification testing while paving the way for full integration between commissioning deliverables such as FATs and SATs with IQ and OQ.
The process validation lifecycle
In 2011, the FDA published “Guidance for Industry: Process Validation: General Principles and Practice.”1 In this guidance (Figure 4), the FDA adopted a lifecycle approach, moving from process qualification to validation in three stages:
- Process Design: In this stage, the commercial manufacturing process is defined based on knowledge gained through development and scale-up activities.
- Process Qualification: In this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing. There are two “substages” or “elements.”
- Design of the facility and qualification of the equipment and utilities; this element refers to all C&Q activities applied to facilities, utilities, systems and equipment.
- Process Performance Qualification (PPQ); this element refers to the traditional “three successful/successive batches for process validation.”
- Continued Process Verification (CPV): There is ongoing assurance that the process remains in a state of control during routine production. This includes routine monitoring of process parameters, trending of data, change control, retraining, and corrective and preventive actions (CAPA), in addition to the periodic review of maintenance and calibration records at pre-established frequencies (based on risk and system classification). For sterile product manufacturing operations, the annual requalification of critical systems (such as autoclaves, SIP, etc.) and semiannual revalidation/re-execution of aseptic process simulations (media fill, for example) is also part of this Stage 3 CPV.
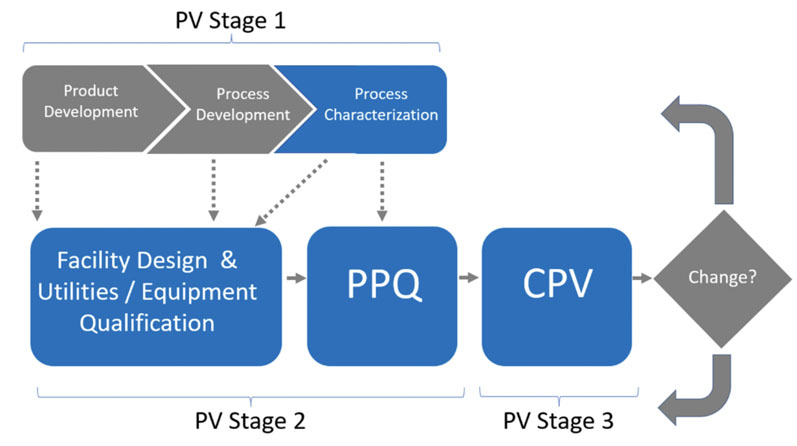
Figure 4: FDA’s process validation model1
Four years later, in 2015, “Annex 15: Qualification & Validation” was published as part of the EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use.2
The following year, EMA published two process validation guidelines.10,11 These used a similar lifecycle approach to the one presented by the FDA. The differences mainly concern terminology:
- the early stages of product and process design are identified by the FDA as “Stage 1, Process Design,” whereas the EU refers to ICH Q89
- Stage 2 is called “Process Qualification (PQ)” by the FDA, whereas the EU document refers to this as “Qualification and Validation”1,2
- in Stage 2, commissioning is included as part of the early activities (the “C” in C&Q); the term “commissioning” is not explicitly referenced in the EU document1,2
- FDA uses the term “qualification” whereas the EU document talks in more granular terms by referring to IQ, OQ and PQ1,2
- what the FDA calls “PPQ,” the EU documents still refer to as “Process Validation”8,9
- Stage 3 is called “Continued Process Verification (CPV)” by the FDA, whereas the EU documents refer to this as “Ongoing Process Verification (OPV).”1,2
Combining them all: the CQV operation lifecycle model
As proposed in Figure 5, a fully integrated CQV operational lifecycle moel is applied to the validation of FUSEP.
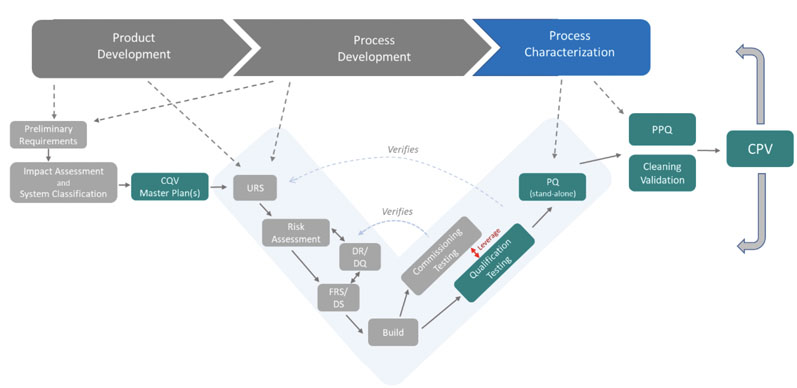
Figure 5: The CQV operational lifecycle model
This model, however, is not applied in the same way for every type of validation. From a CQV planning and project management perspective, the total number of deliverables in a CQV project for a new bio/pharmaceutical facility housing DS and/or DP operations should be divided into five main groups or workstreams (blue squares in Figure 6).
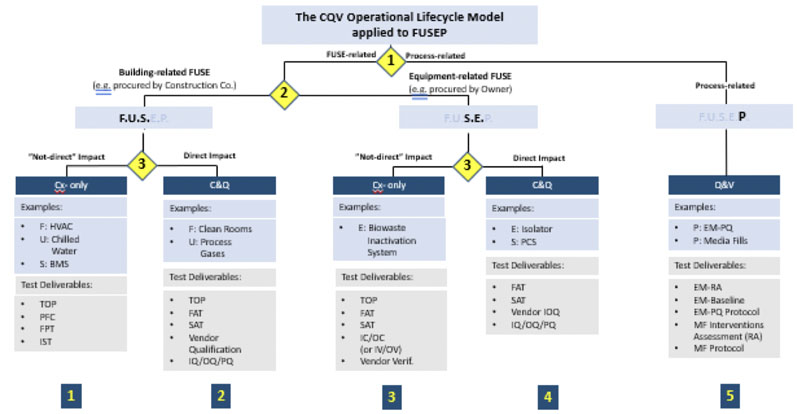
Figure 6: Application of the CQV operational lifecycle model to facilities, utilities, systems, equipment and processes (FUSEP)
The CQV operational lifecycle model can be applied directly to a new biopharmaceutical facility housing both DS and DP operations. There are three distinct decision points based on the following criteria (yellow diamonds in Figure 6):
- Decision Point #1: testing and release of facilities, utilities, systems and equipment (FUSE) versus performance and PPQ testing
- Decision Point #2: testing and release of FUSE systems needed for the start-up and mechanical completion of the facility, usually procured by the construction (building) company versus equipment and systems procured by the owner (when qualification may be completed after mechanical completion)
- Decision Point #3: Extent of testing (commissioning only versus commissioning and qualification) based on impact on product quality (direct versus non-direct).
Guiding the CQV programme
An all-encompassing integrated CQV model can guide the CQV programme in all possible scenarios — whether during the start-up of a new or modified facility or during the performance qualification and continued verification stages.
Although experience has shown that choosing the right validation model can be a complex endeavour, every department within an organisation should work toward this objective.
Once a static process, validation has evolved to become a more dynamic one as pharmaceutical companies seek to simplify validation. They can conduct a risk assessment on their facilities, utilities and equipment to identify whether they impact processes or products.
References
- www.fda.gov/files/drugs/published/Process-Validation--General-Principles-and-Practices.pdf.
- https://health.ec.europa.eu/system/files/2016-11/2015-10_annex15_0.pdf.
- https://ispe.org/initiatives/regulatory/what-gamp#.
- https://ispe.org/sites/default/files/attachments/public/July-Aug-2004.pdf.
- https://www.astm.org/e2500-20.html.
- https://ispe.org/publications/guidance-documents/baseline-guide-vol-5-commissioning-qualification-2nd-edition.
- https://www.ema.europa.eu/en/ich-q9-quality-risk-management-scientific-guideline.
- https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf.
- www.fda.gov/media/77391/download.
- www.ema.europa.eu/en/documents/scientific-guideline/guideline-process-validation-finished-products-information-data-be-provided-regulatory-submissions_en.pdf.
- www.ema.europa.eu/en/documents/scientific-guideline/guideline-process-validation-manufacture-biotechnology-derived-active-substances-data-be-provided_en.pdf.